

Nitric-oxide synthase trafficking inducer is a pleiotropic regulator of endothelial cell function and signaling
- PMID: 28235804
- PMCID: PMC5399110
- DOI: 10.1074/jbc.M116.742627
Nitric-oxide synthase trafficking inducer is a pleiotropic regulator of endothelial cell function and signaling
Abstract
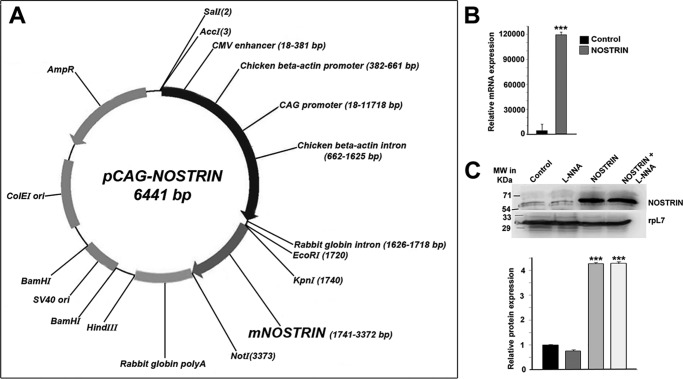
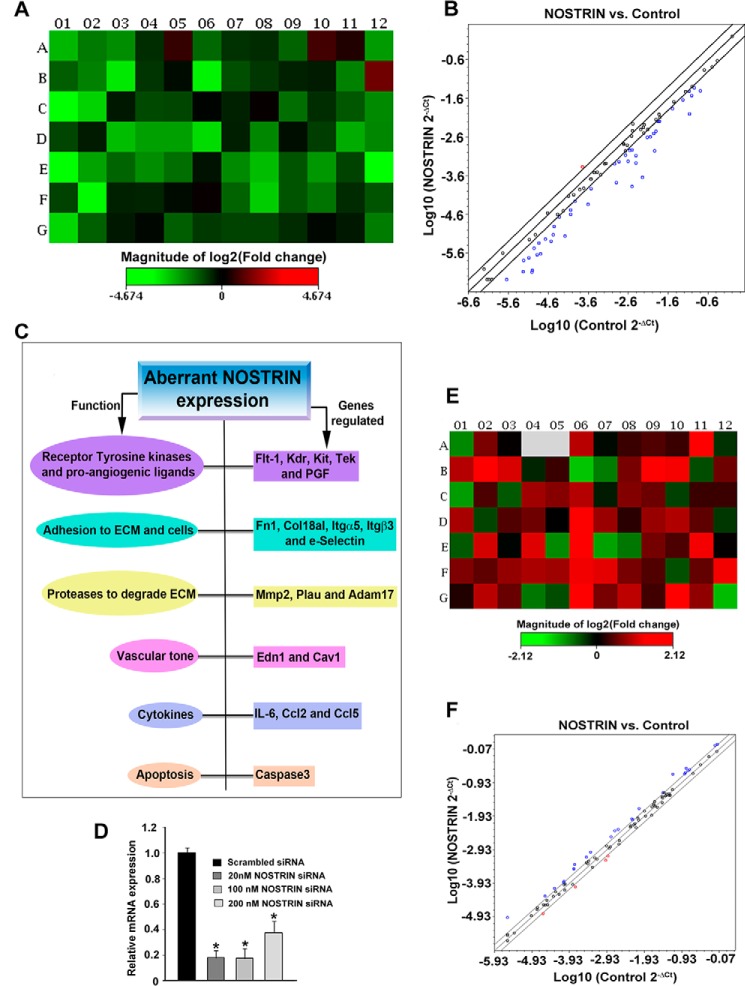
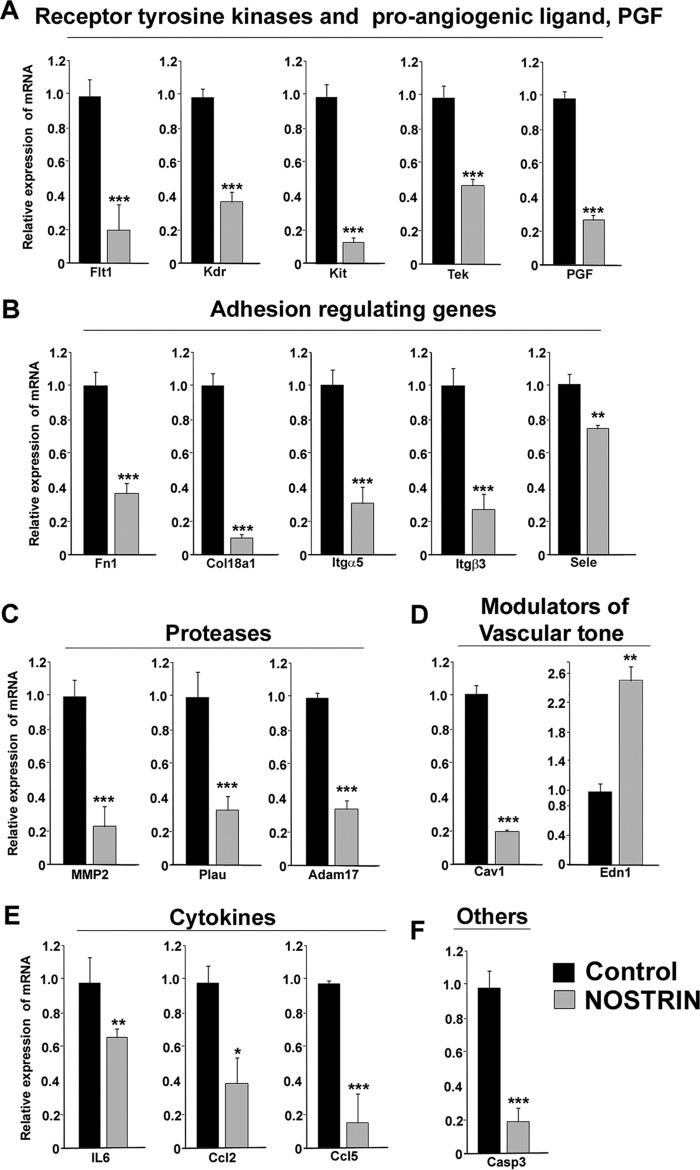
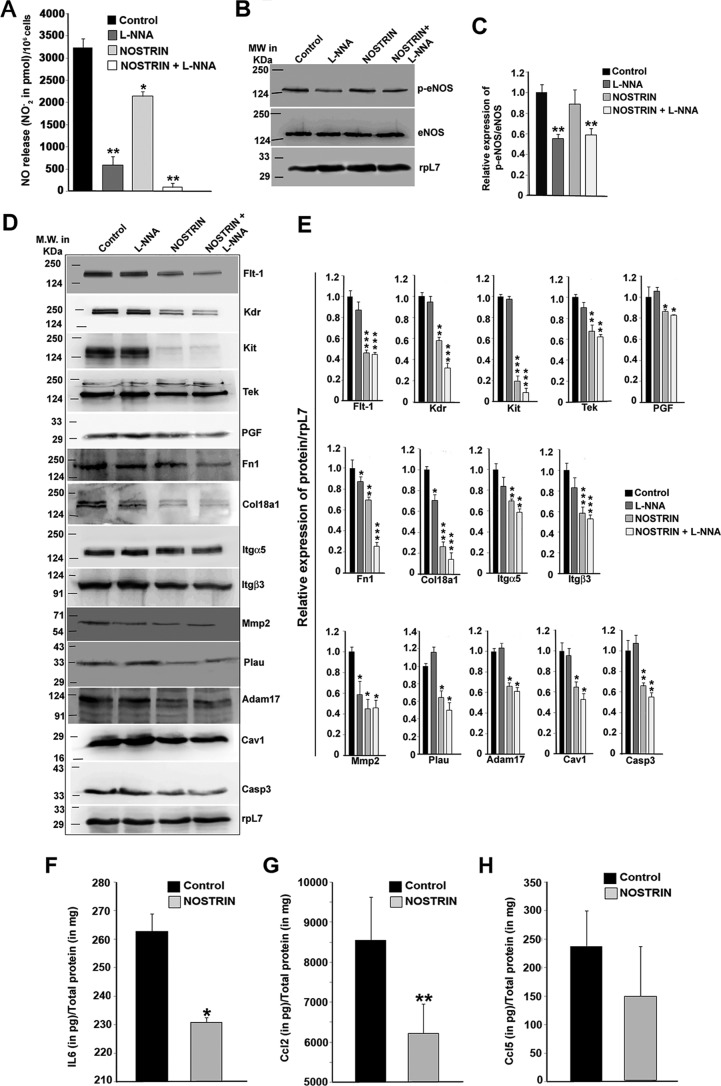
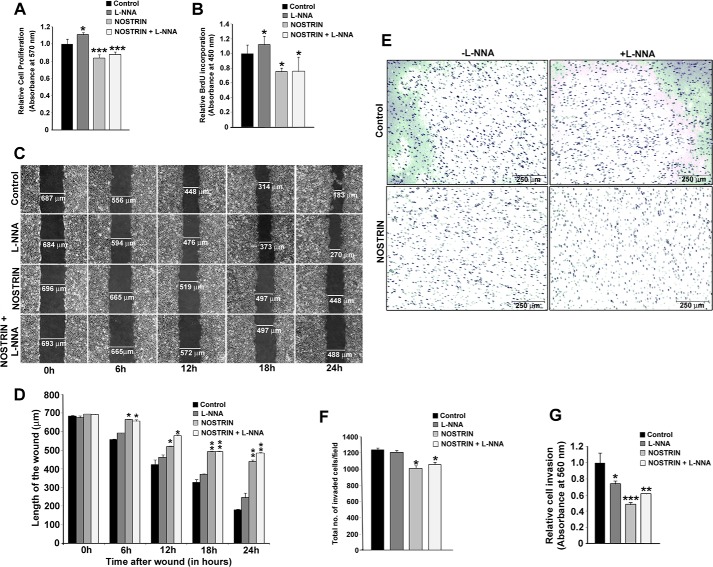
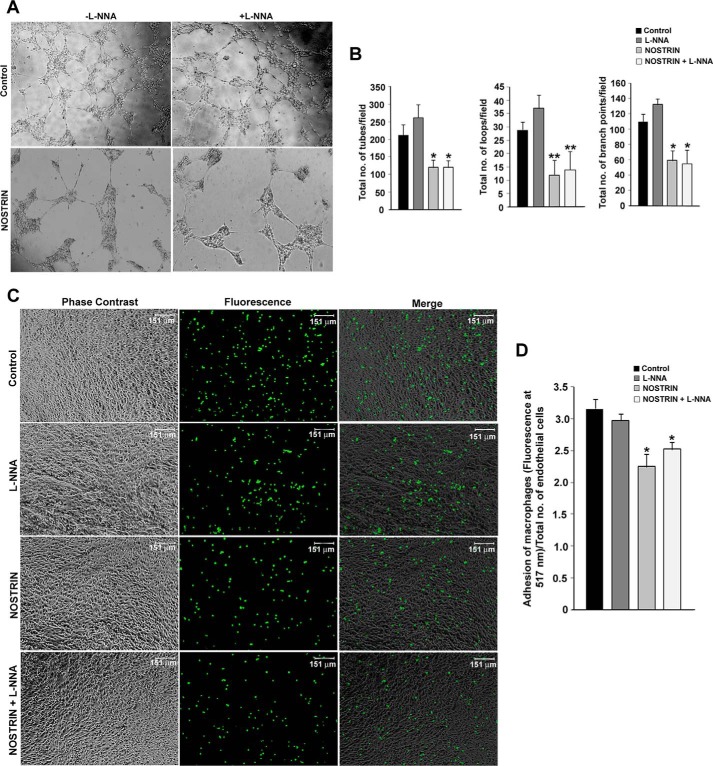
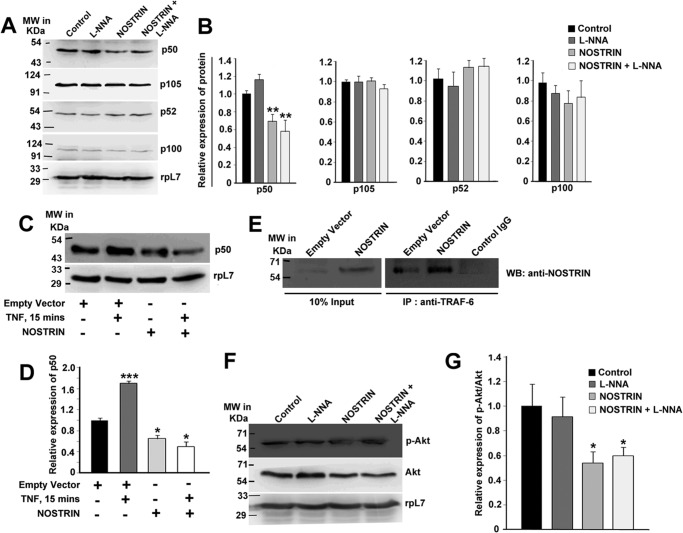
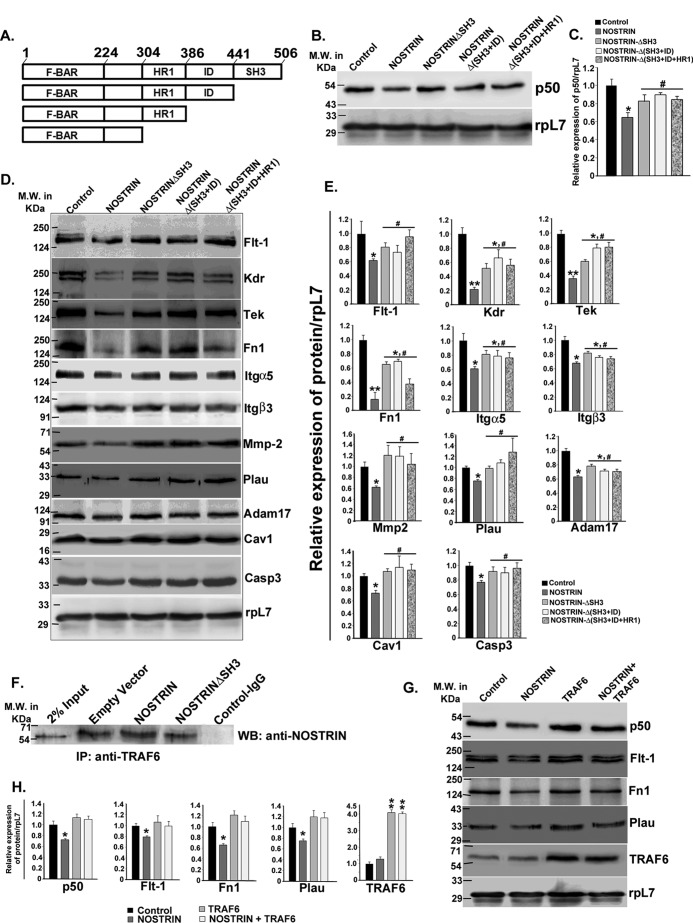
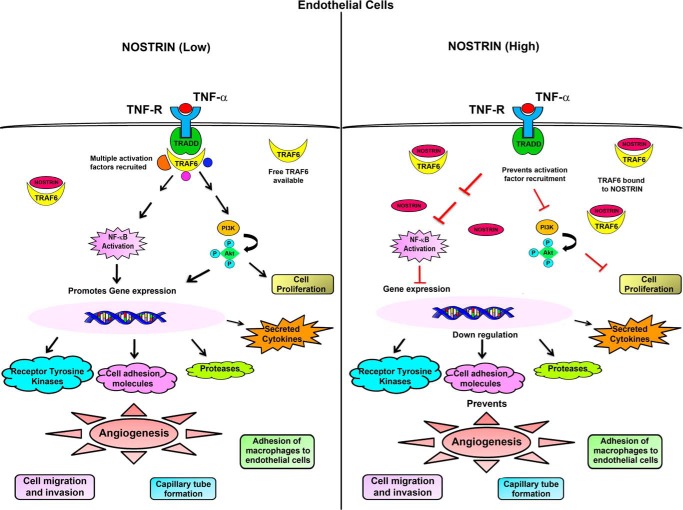
Endothelial nitric-oxide synthase (eNOS) and its bioactive product, nitric oxide (NO), mediate many endothelial cell functions, including angiogenesis and vascular permeability. For example, vascular endothelial growth factor (VEGF)-mediated angiogenesis is inhibited upon reduction of NO bioactivity both in vitro and in vivo Moreover, genetic disruption or pharmacological inhibition of eNOS attenuates angiogenesis during tissue repair, resulting in delayed wound closure. These observations emphasize that eNOS-derived NO can promote angiogenesis. Intriguingly, eNOS activity is regulated by nitric-oxide synthase trafficking inducer (NOSTRIN), which sequesters eNOS, thereby attenuating NO production. This has prompted significant interest in NOSTRIN's function in endothelial cells. We show here that NOSTRIN affects the functional transcriptome of endothelial cells by down-regulating several genes important for invasion and angiogenesis. Interestingly, the effects of NOSTRIN on endothelial gene expression were independent of eNOS activity. NOSTRIN also affected the expression of secreted cytokines involved in inflammatory responses, and ectopic NOSTRIN overexpression functionally restricted endothelial cell proliferation, invasion, adhesion, and VEGF-induced capillary tube formation. Furthermore, NOSTRIN interacted directly with TNF receptor-associated factor 6 (TRAF6), leading to the suppression of NFκB activity and inhibition of AKT activation via phosphorylation. Interestingly, TNF-α-induced NFκB pathway activation was reversed by NOSTRIN. We found that the SH3 domain of NOSTRIN is involved in the NOSTRIN-TRAF6 interaction and is required for NOSTRIN-induced down-regulation of endothelial cell proteins. These results have broad biological implications, as aberrant NOSTRIN expression leading to deactivation of the NFκB pathway, in turn triggering an anti-angiogenic cascade, might inhibit tumorigenesis and cancer progression.
Keywords: TNF receptor-associated factor (TRAF); angiogenesis; endothelial cell; invasion; nitric-oxide synthase.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Carmeliet P. (2003) Angiogenesis in health and disease. Nat. Med. 9, 653–660 - PubMed
-
- Chakraborty S., and Ain R. (2016) Endothelial cell biology: assessment of endothelial cell function, in Trends in Experimental Biology, pp. 1–21, Excel India Publishers, New Delhi, India
-
- Adams R. H., and Alitalo K. (2007) Molecular regulation of angiogenesis and lymph-angiogenesis. Nat. Rev. Mol. Cell Biol. 8, 464–478 - PubMed
-
- De Caterina R., Libby P., Peng H. B., Thannickal V. J., Rajavashisth T. B., Gimbrone M. A. Jr., Shin W. S., and Liao J. K. (1995) Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Invest. 96, 60–68 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
