

Iterative Modeling Reveals Evidence of Sequential Transcriptional Control Mechanisms
- PMID: 28237795
- PMCID: PMC5434763
- DOI: 10.1016/j.cels.2017.01.012
Iterative Modeling Reveals Evidence of Sequential Transcriptional Control Mechanisms
Abstract
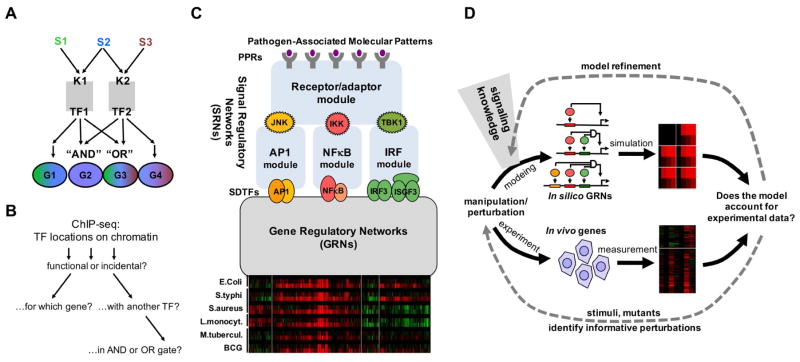
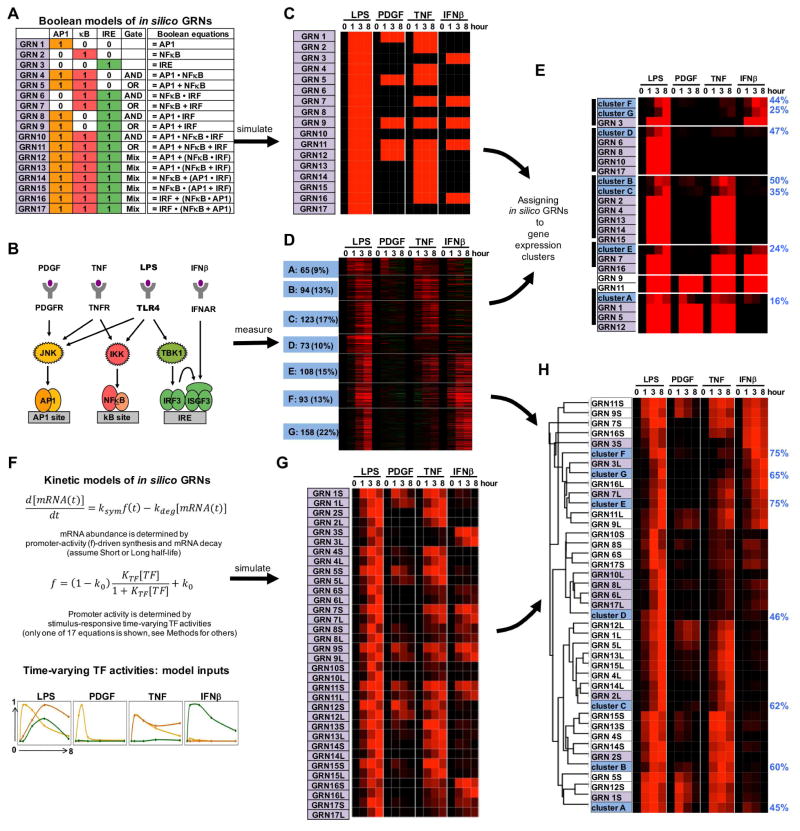
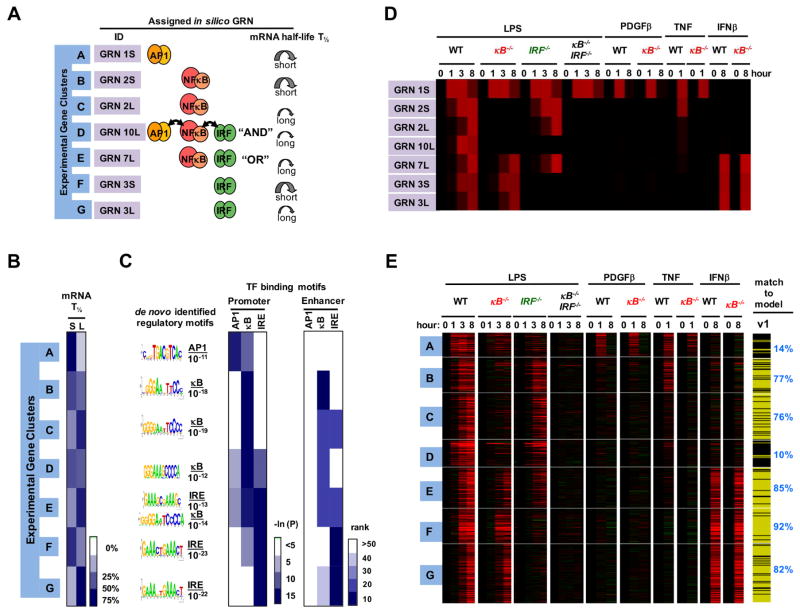
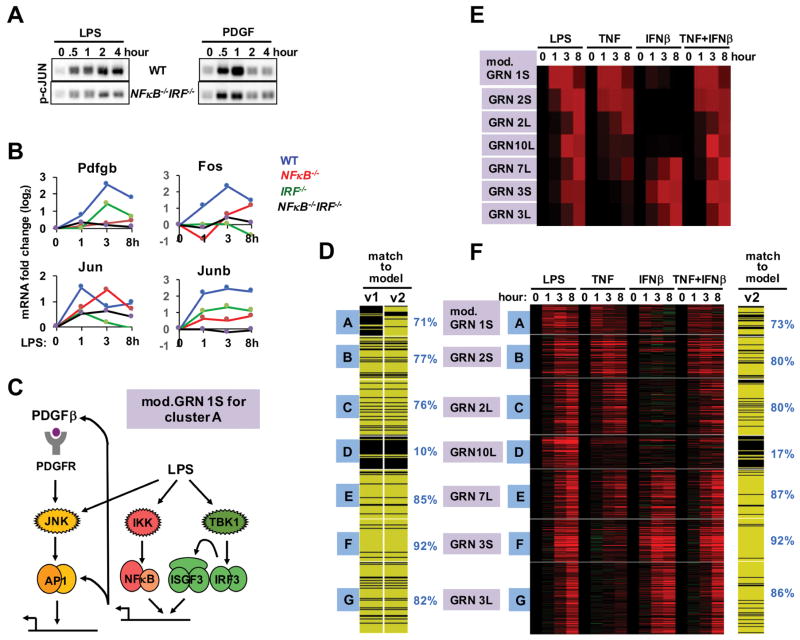
Combinatorial control of gene expression is presumed to be mediated by molecular interactions between coincident transcription factors (TFs). While information on the genome-wide locations of TFs is available, the genes they regulate and whether they function combinatorially often remain open questions. Here, we developed a mechanistic, rather than statistical, modeling approach to elucidate TF control logic from gene expression data. Applying this approach to hundreds of genes in 85 datasets measuring the transcriptional responses of murine fibroblasts and macrophages to cytokines and pathogens, we found that stimulus-responsive TFs generally function sequentially in logical OR gates or singly. Logical AND gates were found between NF-κB-responsive mRNA synthesis and MAPKp38-responsive control of mRNA half-life, but not between temporally coincident TFs. Our analyses identified the functional target genes of each of the pathogen-responsive TFs and prompt a revision of the conceptual underpinnings of combinatorial control of gene expression to include sequentially acting molecular mechanisms that govern mRNA synthesis and decay.
Keywords: combinatorial control; gene expression; gene regulatory logic; mathematical modeling; pathogen responses.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
