

Case Reports
doi: 10.1007/s00401-017-1689-7.
Epub 2017 Feb 25.
Genetic confirmation that ependymoma can arise as part of multiple endocrine neoplasia type 1 (MEN1) syndrome
Affiliations
- PMID: 28238068
- PMCID: PMC5391795
- DOI: 10.1007/s00401-017-1689-7
Item in Clipboard
Case Reports
Genetic confirmation that ependymoma can arise as part of multiple endocrine neoplasia type 1 (MEN1) syndrome
Acta Neuropathol.
2017 Apr.
No abstract available
Figures
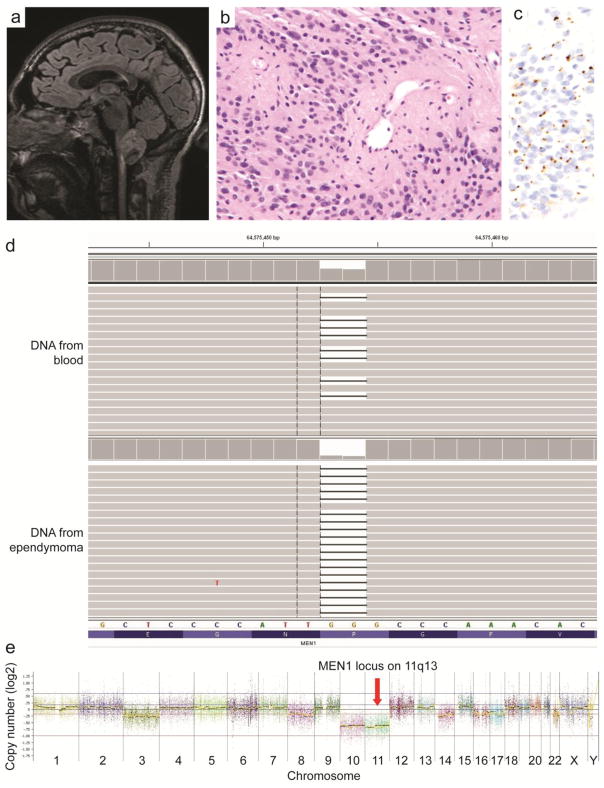
Radiographic, histologic, and genetic features of an ependymoma in a 33 year old man with MEN1 syndrome. a Sagittal T2-weighted fluid-attenuated inversion recovery MRI demonstrating an exophytic, solid and cystic mass arising from the cervicomedullary junction with associated hyperintensity in the superior cervical cord. b H&E stained section of the tumor demonstrating a solid ependymal neoplasm with perivascular pseudorosettes. c Immunostain for EMA demonstrating paranuclear dot-like positivity in tumor cells. d Next-generation sequencing reads for the MEN1 gene demonstrating a 2 base pair deletion (c.563_564delGG, p.P188fs based on transcript NM_130799) present in approximately half of the reads in DNA extracted from the peripheral blood and nearly all of the reads in DNA extracted from the ependymoma. e Genome-wide copy number plot for the tumor demonstrating loss of chromosome 11, which includes the remaining wild-type MEN1 allele at 11q13. Clonal losses of chromosomes 10 and 11 are seen suggesting that they were early events in tumor development, and additional subclonal losses of chromosomes 3, 8, 14, 16, 17, and 22 are also seen that likely occurred later during tumor progression in only a subset of the ependymoma cells.
Similar articles
-
[Case Report No.143: one case of multiple endosecretory-adenomatosis accompanied with ventricula ependymoma].Clin Calcium. 2004 Oct;14(10):150-3. Clin Calcium. 2004. PMID: 15577150 Japanese. No abstract available.
-
Intracranial ependymoma associated with multiple endocrine neoplasia type 1.J Endocrinol Invest. 2010 May;33(5):353-6. doi: 10.1007/BF03346599. Epub 2010 Feb 5. J Endocrinol Invest. 2010. PMID: 20142633 No abstract available.
-
Multiple endocrine neoplasia type 1 associated with spinal ependymoma.Intern Med. 1996 Apr;35(4):285-9. doi: 10.2169/internalmedicine.35.285. Intern Med. 1996. PMID: 8739783 Review.
-
A novel mutation of the MEN1 gene in a patient with multiple endocrine neoplasia type 1 and recurrent fibromyxoid sarcoma - a case report.BMC Med Genet. 2020 Sep 29;21(1):190. doi: 10.1186/s12881-020-01129-4. BMC Med Genet. 2020. PMID: 32993530 Free PMC article.
-
[Multiple endocrine neoplasia type 1 variants and phenocopies].Ter Arkh. 2014;86(10):87-91. Ter Arkh. 2014. PMID: 25509899 Review. Russian.
Cited by
-
Beyond the "3 Ps": A critical appraisal of the non-endocrine manifestations of multiple endocrine neoplasia type 1.Front Endocrinol (Lausanne). 2022 Oct 17;13:1029041. doi: 10.3389/fendo.2022.1029041. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36325452 Free PMC article. Review.
-
Childhood Multiple Endocrine Neoplasia (MEN) Syndromes: Genetics, Clinical Heterogeneity and Modifying Genes.J Clin Med. 2024 Sep 18;13(18):5510. doi: 10.3390/jcm13185510. J Clin Med. 2024. PMID: 39336996 Free PMC article. Review.
-
Clinical aspects of multiple endocrine neoplasia type 1.Nat Rev Endocrinol. 2021 Apr;17(4):207-224. doi: 10.1038/s41574-021-00468-3. Epub 2021 Feb 9. Nat Rev Endocrinol. 2021. PMID: 33564173 Review.
-
The DNA methylome of pediatric brain tumors appears shaped by structural variation and predicts survival.Nat Commun. 2024 Aug 8;15(1):6775. doi: 10.1038/s41467-024-51276-y. Nat Commun. 2024. PMID: 39117669 Free PMC article.
-
PDP type brain tumor in association with multiple endocrine neoplasia type 1.Heliyon. 2024 Mar 12;10(6):e27418. doi: 10.1016/j.heliyon.2024.e27418. eCollection 2024 Mar 30. Heliyon. 2024. PMID: 38510015 Free PMC article.
References
-
- Al-Salameh A, Francois P, Giraud S. Intracranial ependymoma associated with multiple endocrine neoplasia type 1. J Endocrinol Invest. 2010;33:353–356. - PubMed
-
- Funayama T, Sakane M, Yoshizawa T, et al. Tanycytic ependymoma of the filum terminale associated with multiple endocrine neoplasia 1: first reported case. Spine J. 2013;13:e49–54. - PubMed
-
- Giraud S, Choplin H, Teh BT, et al. A large multiple endocrine neoplasia type 1 family with clinical expression suggestive of anticipation. J Clin Endocrinol Metab. 1997;82:3487–3492. - PubMed
-
- Kato H, Uchimura I, Morohoshi M, et al. Multiple endocrine neoplasia type 1 associated with spinal ependymoma. Intern Med. 1996;35:285–289. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
