

Efficient computation of the joint sample frequency spectra for multiple populations
- PMID: 28239248
- PMCID: PMC5319604
- DOI: 10.1080/10618600.2016.1159212
Efficient computation of the joint sample frequency spectra for multiple populations
Abstract
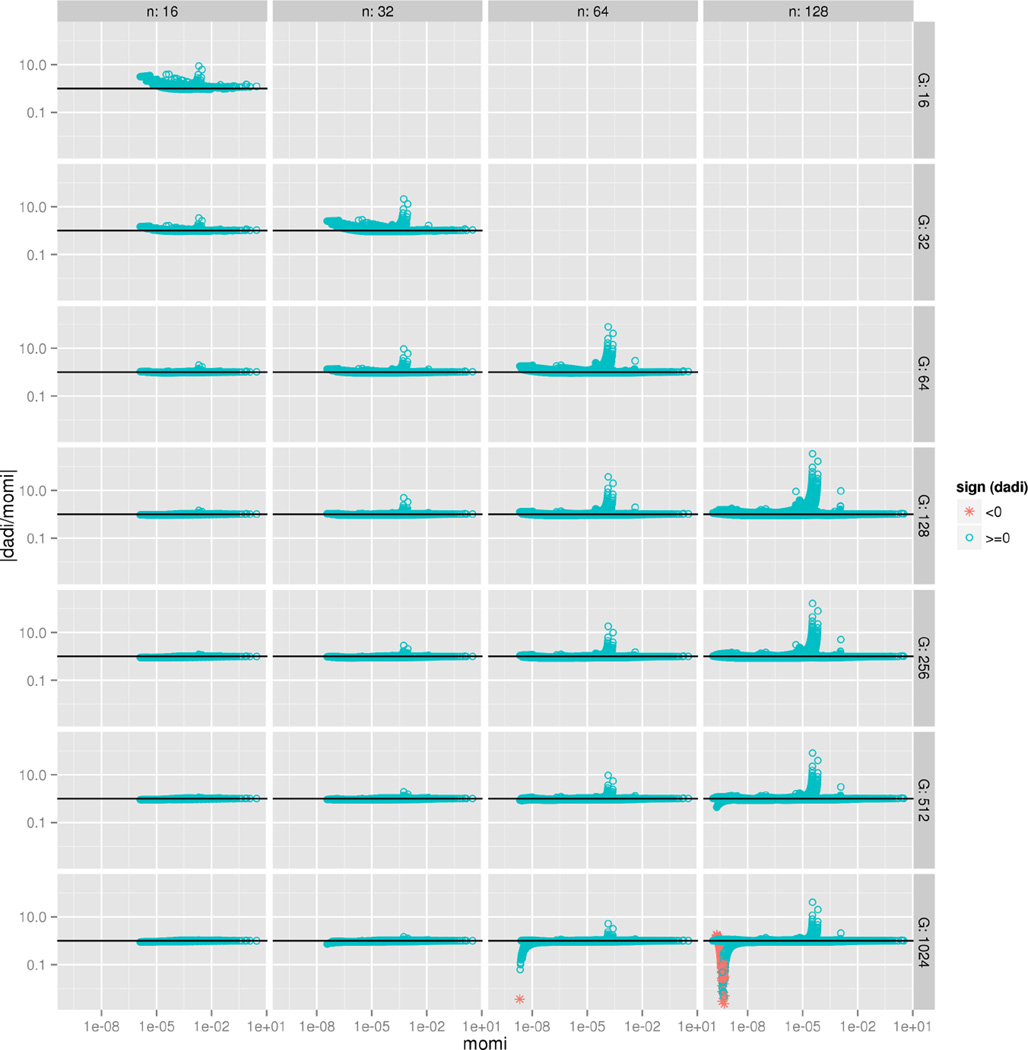
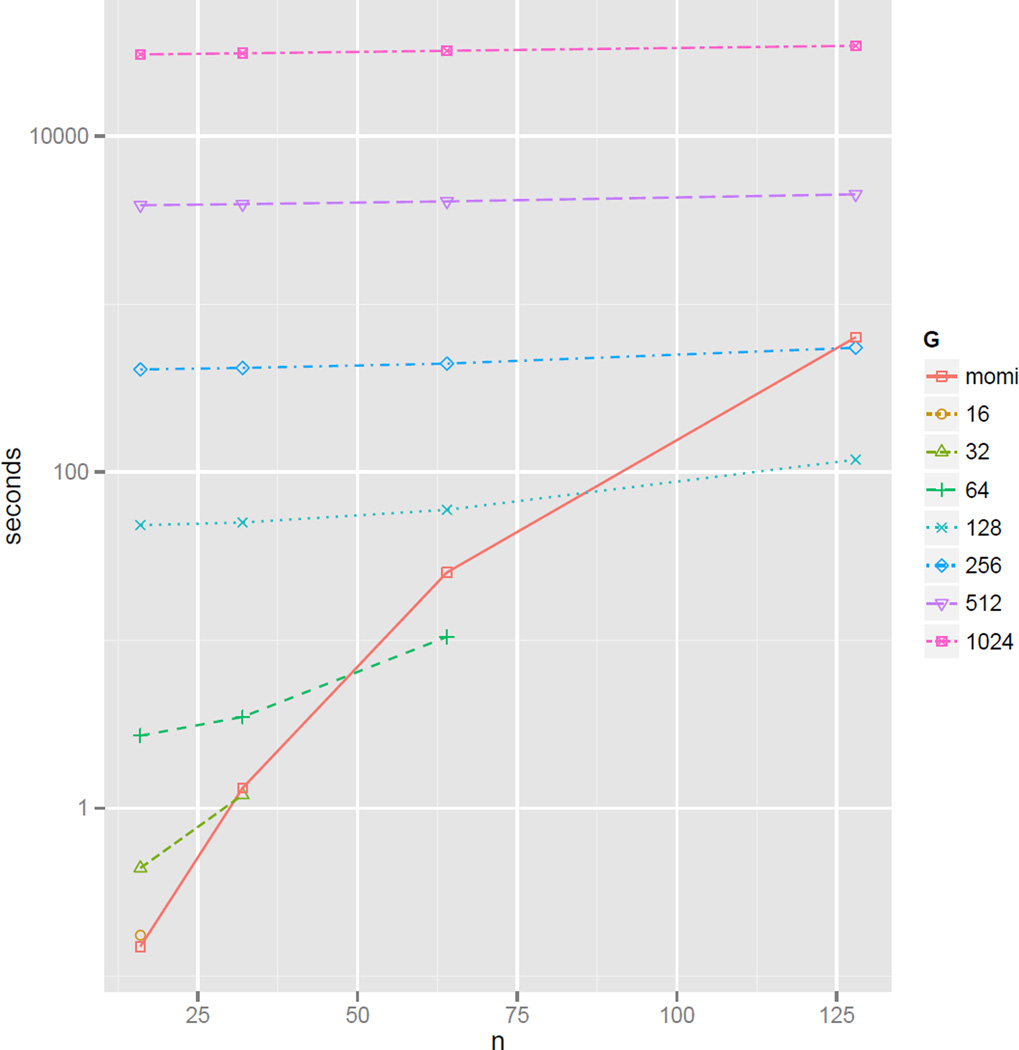
A wide range of studies in population genetics have employed the sample frequency spectrum (SFS), a summary statistic which describes the distribution of mutant alleles at a polymorphic site in a sample of DNA sequences and provides a highly efficient dimensional reduction of large-scale population genomic variation data. Recently, there has been much interest in analyzing the joint SFS data from multiple populations to infer parameters of complex demographic histories, including variable population sizes, population split times, migration rates, admixture proportions, and so on. SFS-based inference methods require accurate computation of the expected SFS under a given demographic model. Although much methodological progress has been made, existing methods suffer from numerical instability and high computational complexity when multiple populations are involved and the sample size is large. In this paper, we present new analytic formulas and algorithms that enable accurate, efficient computation of the expected joint SFS for thousands of individuals sampled from hundreds of populations related by a complex demographic model with arbitrary population size histories (including piecewise-exponential growth). Our results are implemented in a new software package called momi (MOran Models for Inference). Through an empirical study we demonstrate our improvements to numerical stability and computational complexity.
Keywords: coalescent; demographic inference; population genetics; sum-product algorithm.
Figures
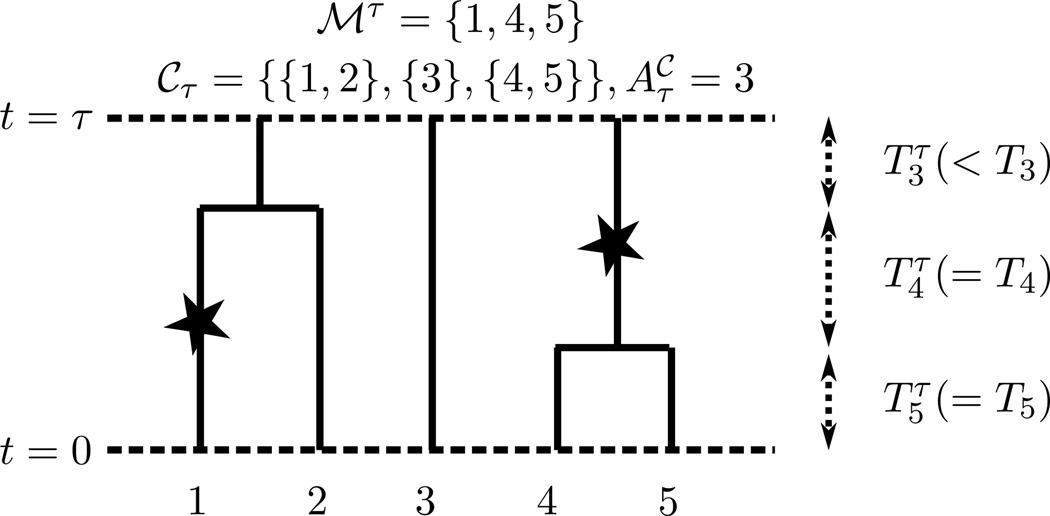
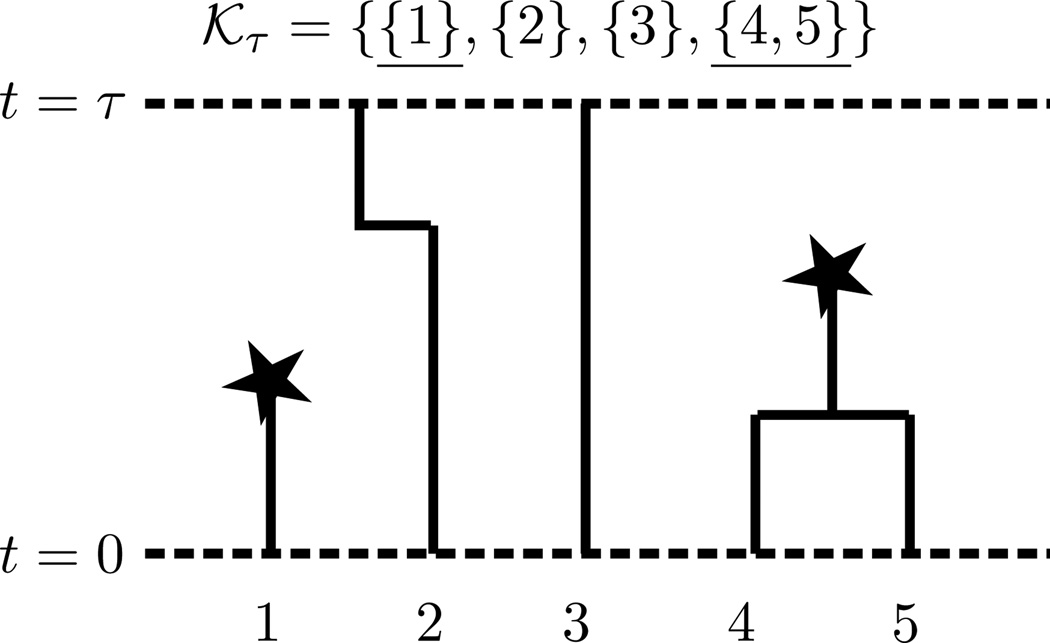
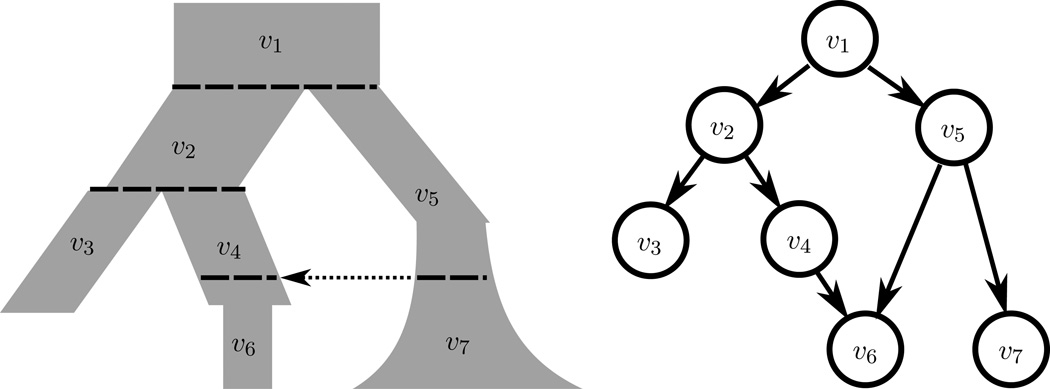

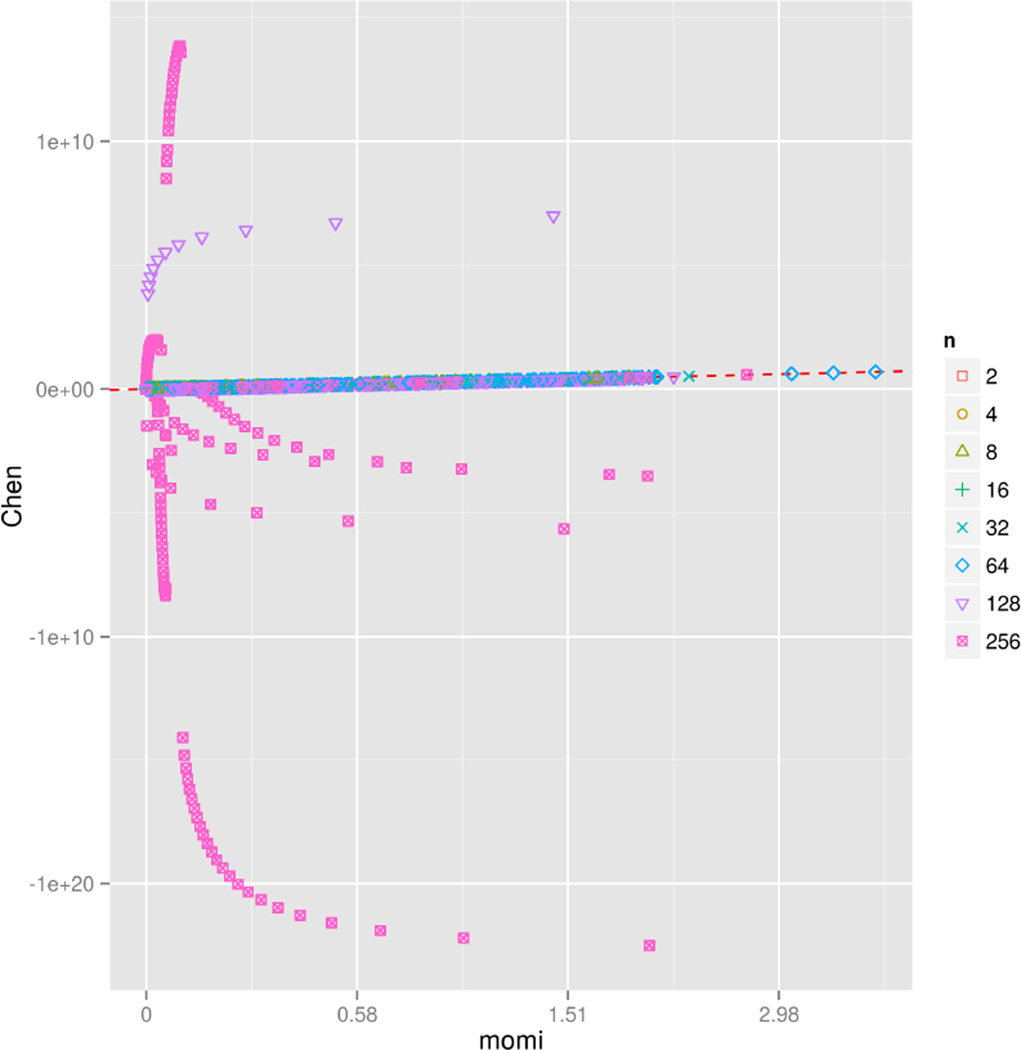
References
-
- Al-Mohy AH, Higham NJ. Computing the action of the matrix exponential, with an application to exponential integrators. SIAM Journal on Scientific Computing. 2011;33(2):488–511.
-
- Aldous DJ. Exchangeability and related topics. In: Hennequin P, editor. École d' Été de Probabilités de Saint-Flour XIII — 1983, volume 1117 of Lecture Notes in Mathematics. Berlin Heidelberg: Springer; 1985. pp. 1–198.
-
- Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London. Series B: Biological Sciences. 1996;263(1377):1619–1626.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources