

Switching harmful visceral fat to beneficial energy combustion improves metabolic dysfunctions
- PMID: 28239649
- PMCID: PMC5313060
- DOI: 10.1172/jci.insight.89044
Switching harmful visceral fat to beneficial energy combustion improves metabolic dysfunctions
Abstract
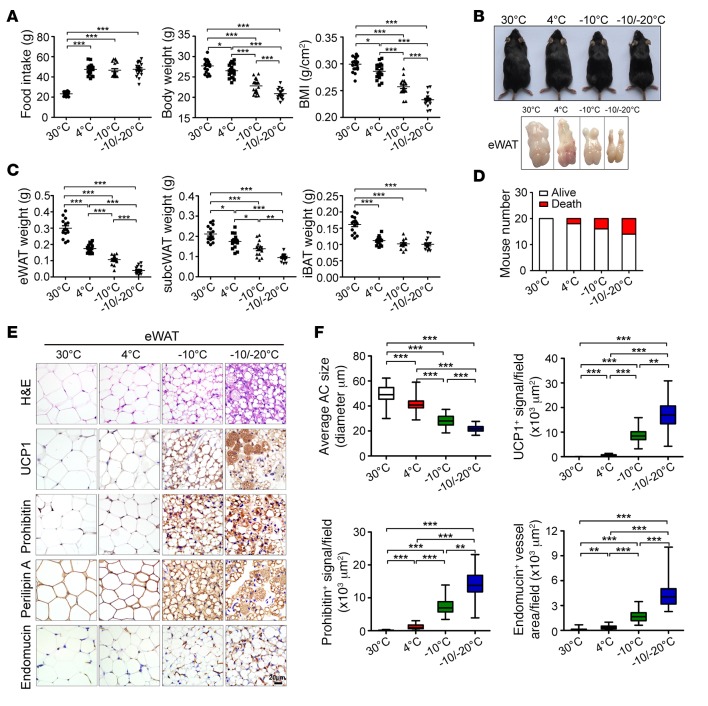
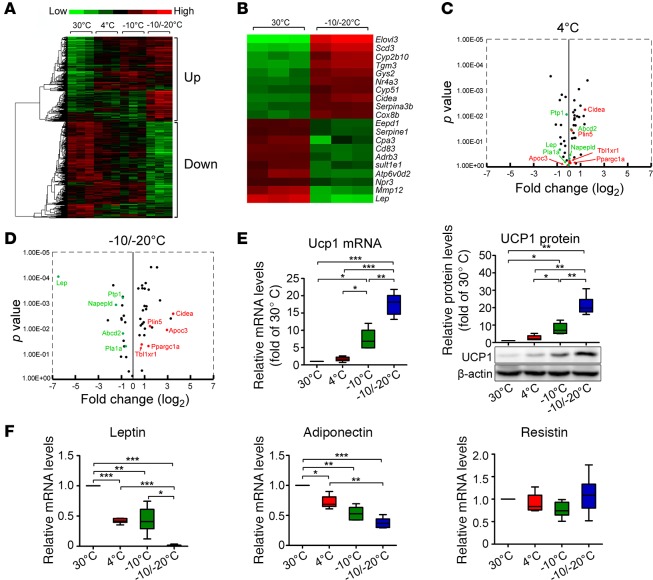
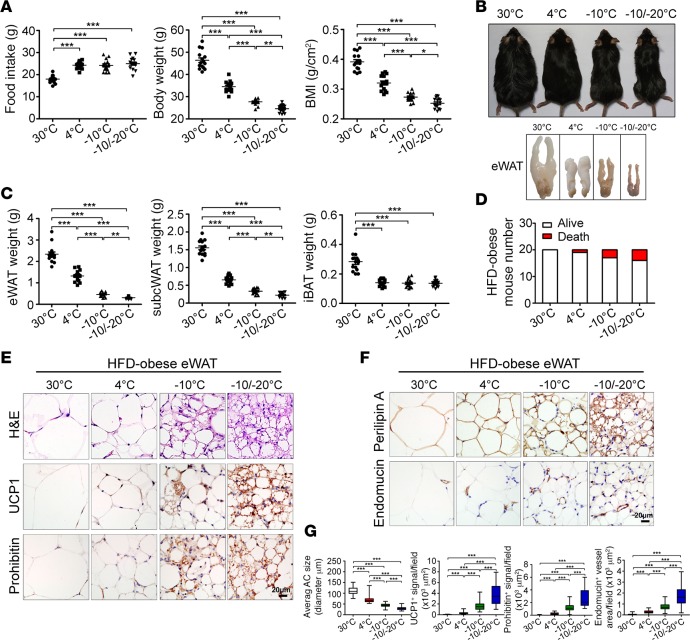
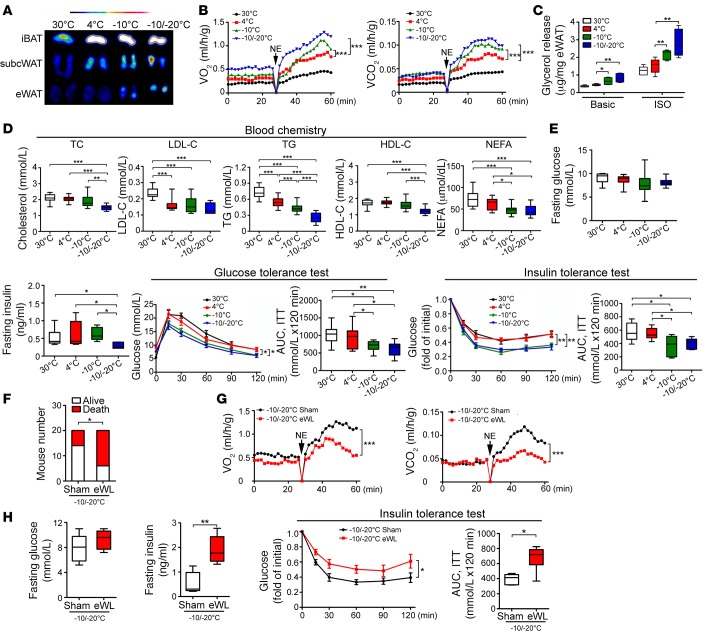
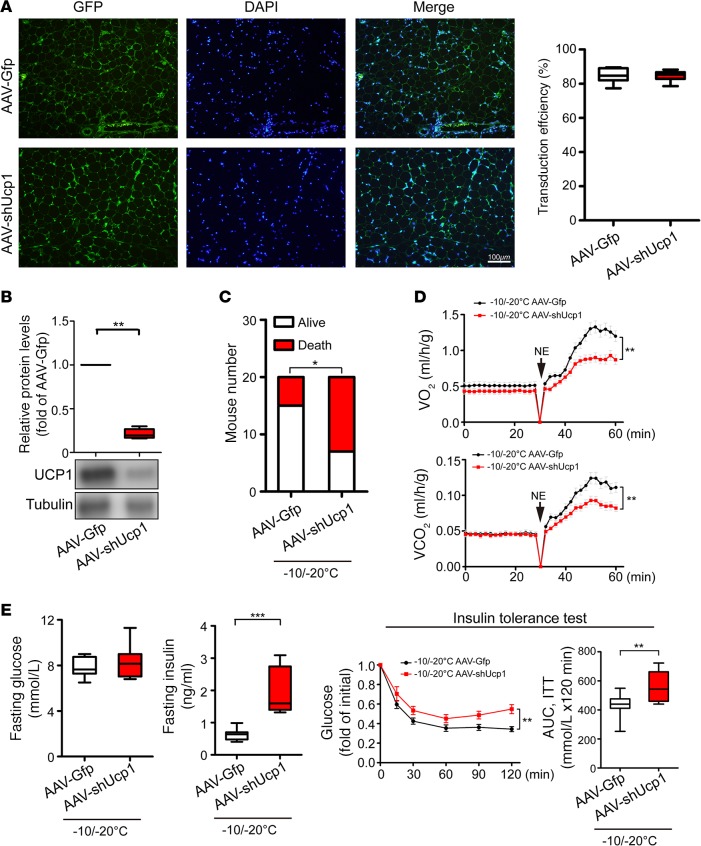
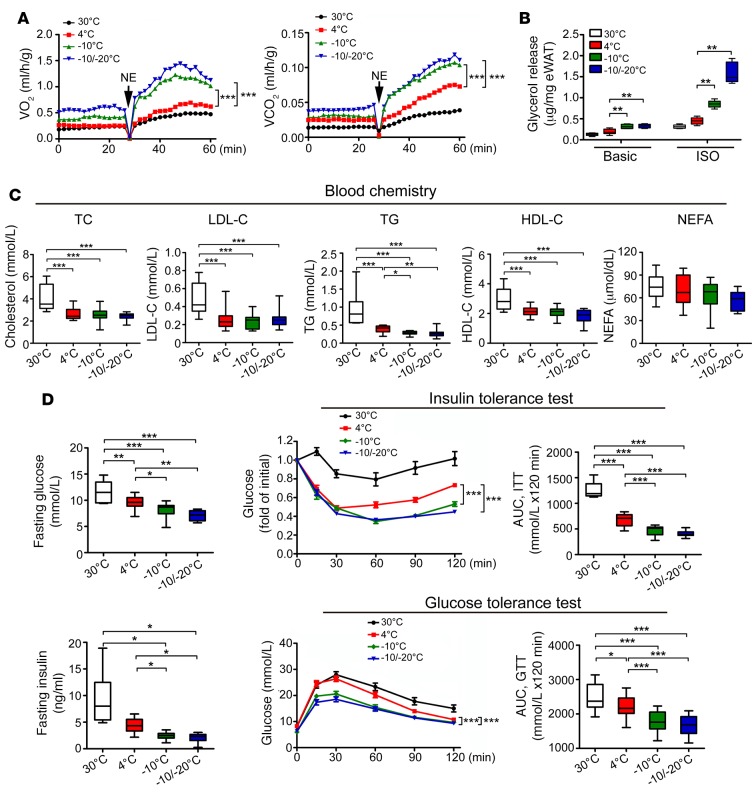
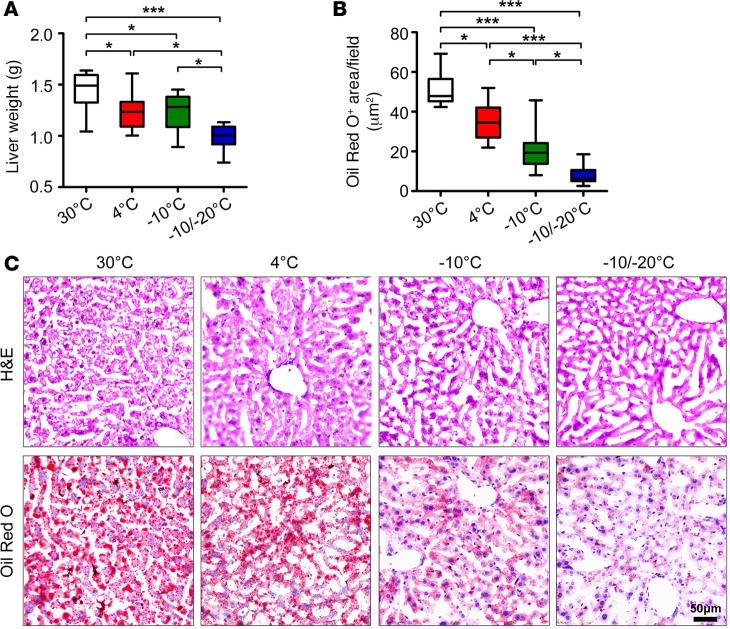
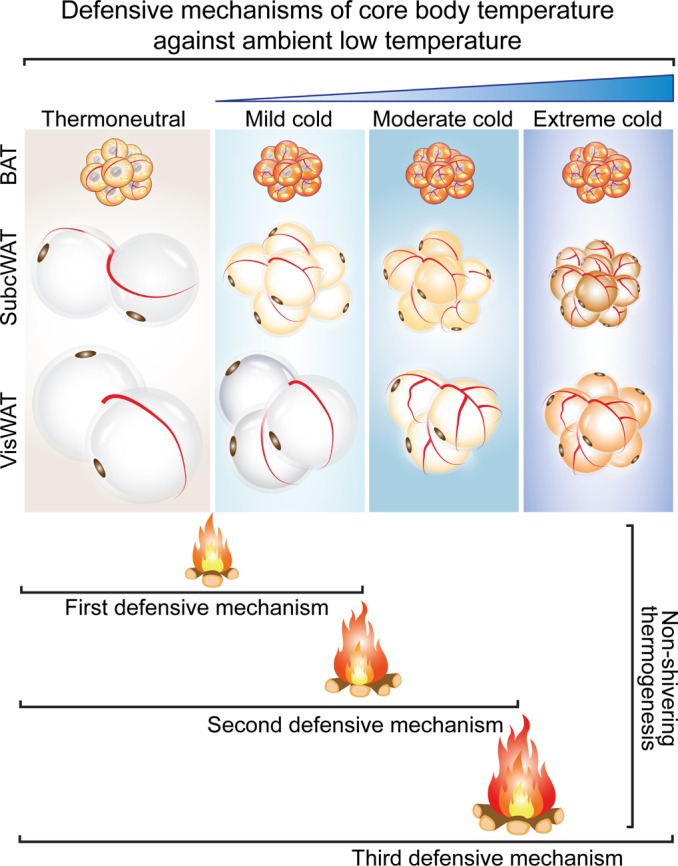
Visceral fat is considered the genuine and harmful white adipose tissue (WAT) that is associated to development of metabolic disorders, cardiovascular disease, and cancer. Here, we present a new concept to turn the harmful visceral fat into a beneficial energy consumption depot, which is beneficial for improvement of metabolic dysfunctions in obese mice. We show that low temperature-dependent browning of visceral fat caused decreased adipose weight, total body weight, and body mass index, despite increased food intake. In high-fat diet-fed mice, low temperature exposure improved browning of visceral fat, global metabolism via nonshivering thermogenesis, insulin sensitivity, and hepatic steatosis. Genome-wide expression profiling showed upregulation of WAT browning-related genes including Cidea and Dio2. Conversely, Prdm16 was unchanged in healthy mice or was downregulated in obese mice. Surgical removal of visceral fat and genetic knockdown of UCP1 in epididymal fat largely ablated low temperature-increased global thermogenesis and resulted in the death of most mice. Thus, browning of visceral fat may be a compensatory heating mechanism that could provide a novel therapeutic strategy for treating visceral fat-associated obesity and diabetes.
Conflict of interest statement
Conflict of interest: The authors have declared that no conflict of interest exists.
Figures
References
-
- Kopelman PG. Obesity as a medical problem. Nature. 2000;404(6778):635–643. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
