

Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects
- PMID: 28240606
- PMCID: PMC5373883
- DOI: 10.1172/JCI89857
Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects
Abstract
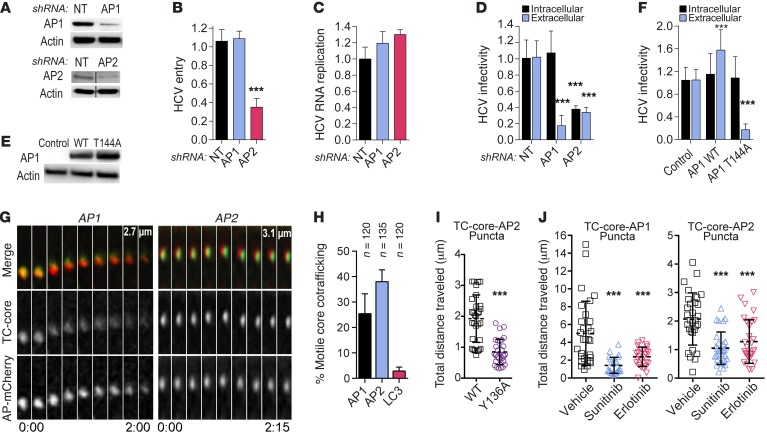
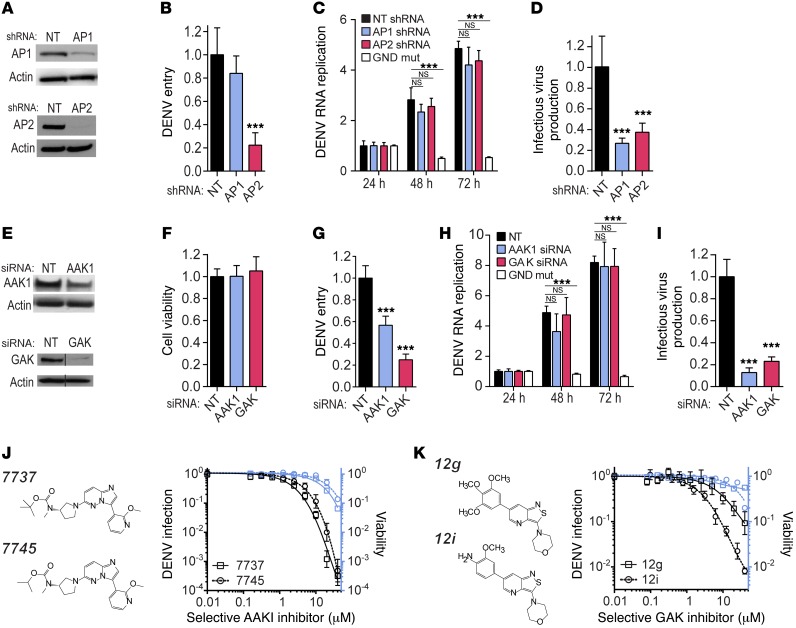
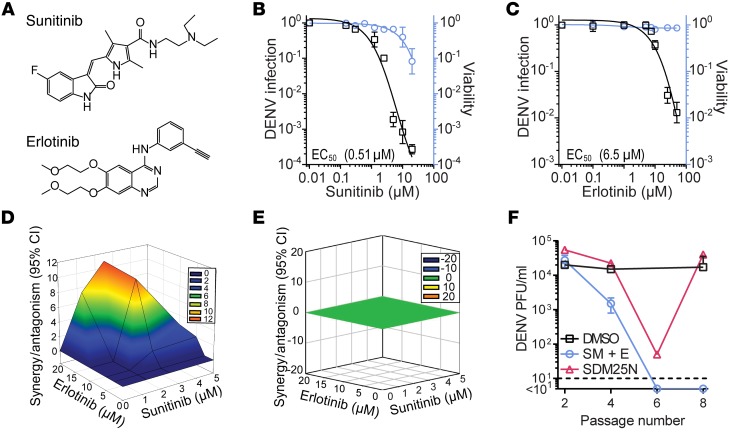
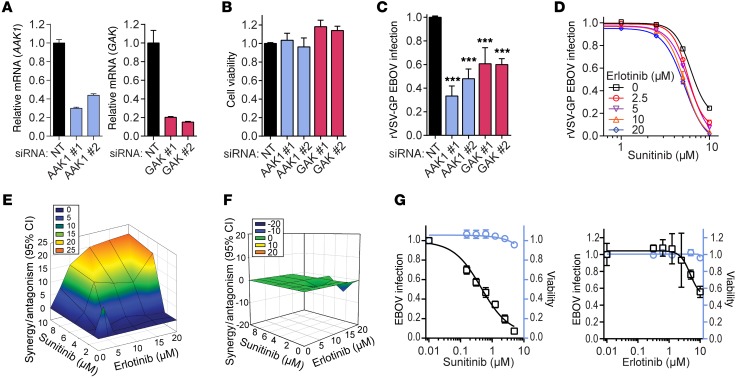
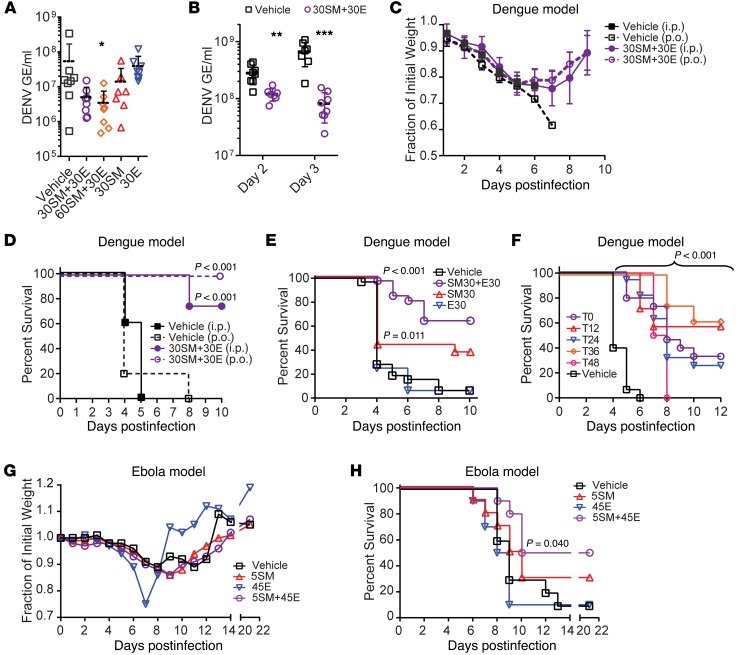
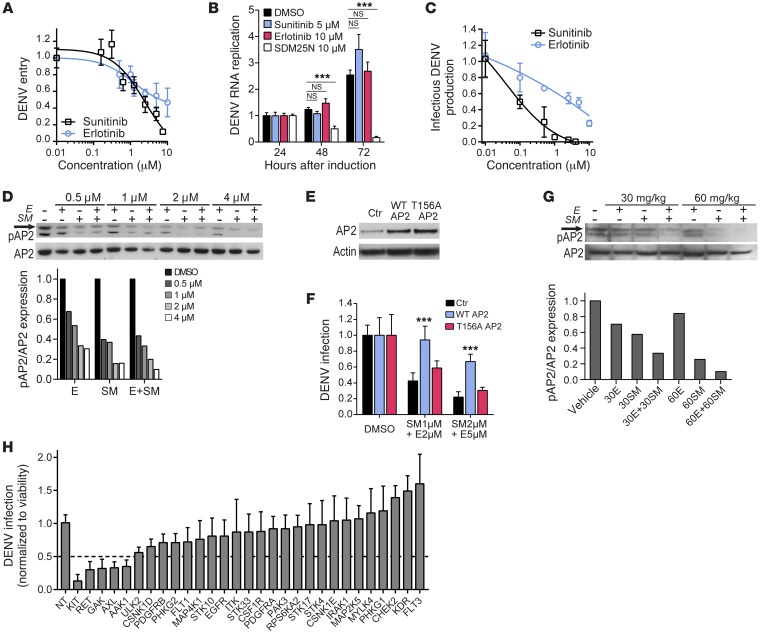
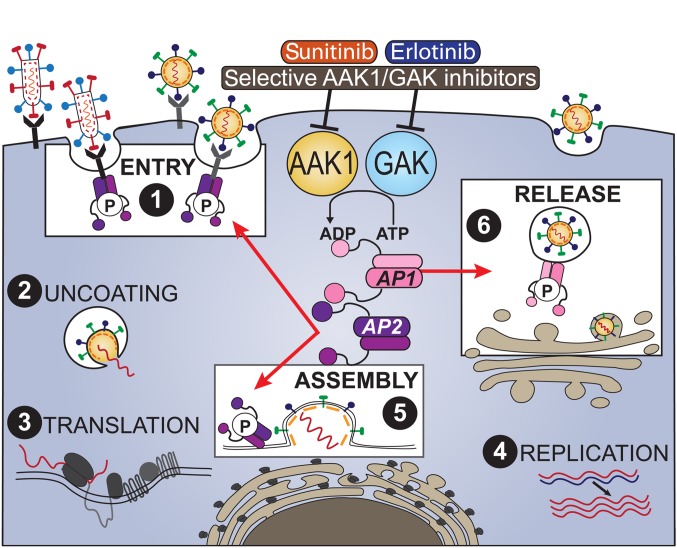
Global health is threatened by emerging viral infections, which largely lack effective vaccines or therapies. Targeting host pathways that are exploited by multiple viruses could offer broad-spectrum solutions. We previously reported that AAK1 and GAK, kinase regulators of the host adaptor proteins AP1 and AP2, are essential for hepatitis C virus (HCV) infection, but the underlying mechanism and relevance to other viruses or in vivo infections remained unknown. Here, we have discovered that AP1 and AP2 cotraffic with HCV particles in live cells. Moreover, we found that multiple viruses, including dengue and Ebola, exploit AAK1 and GAK during entry and infectious virus production. In cultured cells, treatment with sunitinib and erlotinib, approved anticancer drugs that inhibit AAK1 or GAK activity, or with more selective compounds inhibited intracellular trafficking of HCV and multiple unrelated RNA viruses with a high barrier to resistance. In murine models of dengue and Ebola infection, sunitinib/erlotinib combination protected against morbidity and mortality. We validated sunitinib- and erlotinib-mediated inhibition of AAK1 and GAK activity as an important mechanism of antiviral action. Additionally, we revealed potential roles for additional kinase targets. These findings advance our understanding of virus-host interactions and establish a proof of principle for a repurposed, host-targeted approach to combat emerging viruses.
Conflict of interest statement
Figures
Comment in
-
Viral infections: Targeting host kinases.Nat Rev Drug Discov. 2017 May;16(5):314. doi: 10.1038/nrd.2017.75. Epub 2017 Apr 18. Nat Rev Drug Discov. 2017. PMID: 28417985 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
