

BBCAnalyzer: a visual approach to facilitate variant calling
- PMID: 28241736
- PMCID: PMC5330023
- DOI: 10.1186/s12859-017-1549-4
BBCAnalyzer: a visual approach to facilitate variant calling
Abstract
Background: Deriving valid variant calling results from raw next-generation sequencing data is a particularly challenging task, especially with respect to clinical diagnostics and personalized medicine. However, when using classic variant calling software, the user usually obtains nothing more than a list of variants that pass the corresponding caller's internal filters. Any expected mutations (e.g. hotspot mutations), that have not been called by the software, need to be investigated manually.
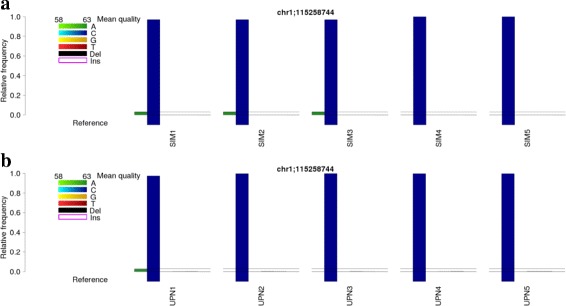
Results: BBCAnalyzer (Bases By CIGAR Analyzer) provides a novel visual approach to facilitate this step of time-consuming, manual inspection of common mutation sites. BBCAnalyzer is able to visualize base counts at predefined positions or regions in any sequence alignment data that are available as BAM files. Thereby, the tool provides a straightforward solution for evaluating any list of expected mutations like hotspot mutations, or even whole regions of interest. In addition to an ordinary textual report, BBCAnalyzer reports highly customizable plots. Information on the counted number of bases, the reference bases, known mutations or polymorphisms, called mutations and base qualities is summarized in a single plot. By uniting this information in a graphical way, the user may easily decide on a variant being present or not - completely independent of any internal filters or frequency thresholds.
Conclusions: BBCAnalyzer provides a unique, novel approach to facilitate variant calling where classical tools frequently fail to call. The R package is freely available at http://bioconductor.org . The local web application is available at Additional file 2. A documentation of the R package (Additional file 1) as well as the web application (Additional file 2) with detailed descriptions, examples of all input- and output elements, exemplary code as well as exemplary data are included. A video demonstrates the exemplary usage of the local web application (Additional file 3). Additional file 3: Supplement_3. Video demonstrating the exemplary usage of the web application "BBCAnalyzer". (MP4 11571 kb).
Keywords: Hotspot mutations; Next-generation sequencing; Personalized medicine; Variant calling; Visualization.
Figures


Similar articles
-
Evaluating alignment and variant-calling software for mutation identification in C. elegans by whole-genome sequencing.PLoS One. 2017 Mar 23;12(3):e0174446. doi: 10.1371/journal.pone.0174446. eCollection 2017. PLoS One. 2017. PMID: 28333980 Free PMC article.
-
AMLVaran: a software approach to implement variant analysis of targeted NGS sequencing data in an oncological care setting.BMC Med Genomics. 2020 Feb 4;13(1):17. doi: 10.1186/s12920-020-0668-3. BMC Med Genomics. 2020. PMID: 32019565 Free PMC article.
-
Targeted Next Generation Sequencing as a Reliable Diagnostic Assay for the Detection of Somatic Mutations in Tumours Using Minimal DNA Amounts from Formalin Fixed Paraffin Embedded Material.PLoS One. 2016 Feb 26;11(2):e0149405. doi: 10.1371/journal.pone.0149405. eCollection 2016. PLoS One. 2016. PMID: 26919633 Free PMC article.
-
Review of alignment and SNP calling algorithms for next-generation sequencing data.J Appl Genet. 2016 Feb;57(1):71-9. doi: 10.1007/s13353-015-0292-7. Epub 2015 Jun 9. J Appl Genet. 2016. PMID: 26055432 Review.
-
vcfView: An Extensible Data Visualization and Quality Assurance Platform for Integrated Somatic Variant Analysis.Cancer Inform. 2020 Nov 11;19:1176935120972377. doi: 10.1177/1176935120972377. eCollection 2020. Cancer Inform. 2020. PMID: 33239857 Free PMC article. Review.
References
-
- DePristo M, Banks E, Poplin R, Garimella K, Maguire J, Hartl C, Philippakis A, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell T, Kernytsky A, Sivachenko A, Cibulskis K, Gabriel S, Altshuler D, Daly M. A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. - DOI - PMC - PubMed
-
- Münz M, Ruark E, Renwick A, Ramsay E, Clarke M, Mahamdallie S, Cloke V, Seal S, Strydom A, Lunter G, Rahman N. CSN and CAVA: variant annotation tools for rapid, robust next-generation sequencing analysis in the clinical setting. Genome Med. 2015;7:1–8. doi: 10.1186/s13073-015-0195-6. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
