

Epilepsy-associated GRIN2A mutations reduce NMDA receptor trafficking and agonist potency - molecular profiling and functional rescue
- PMID: 28242877
- PMCID: PMC5427847
- DOI: 10.1038/s41598-017-00115-w
Epilepsy-associated GRIN2A mutations reduce NMDA receptor trafficking and agonist potency - molecular profiling and functional rescue
Abstract
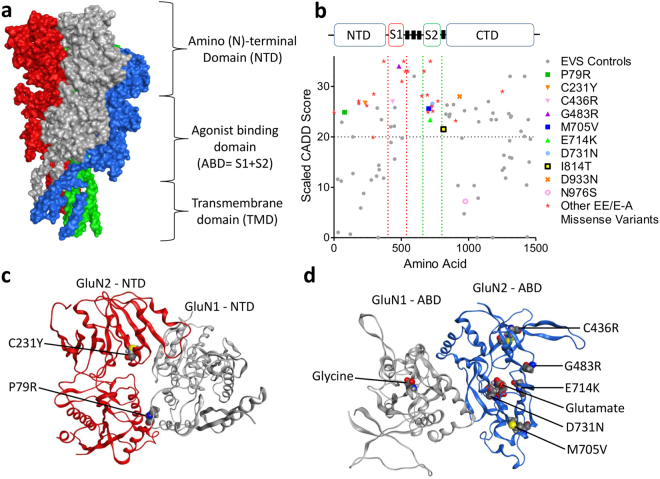
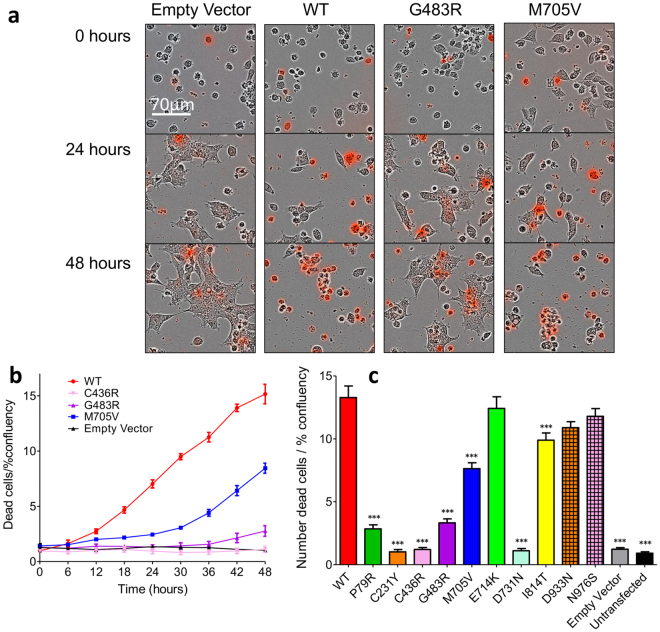
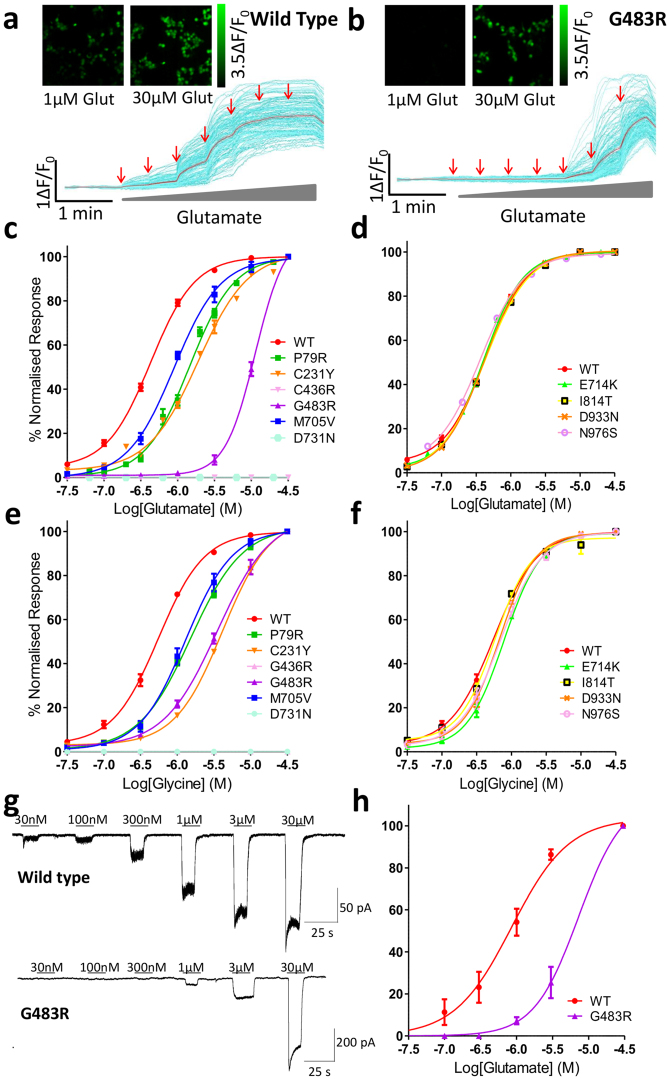
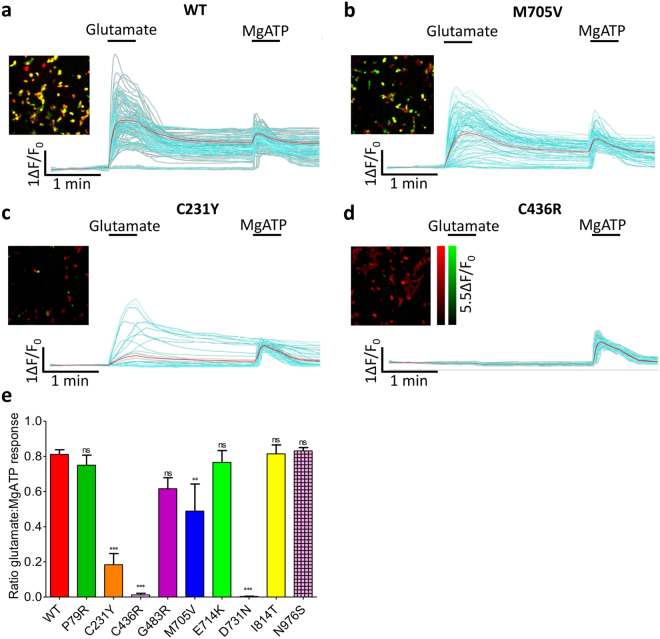
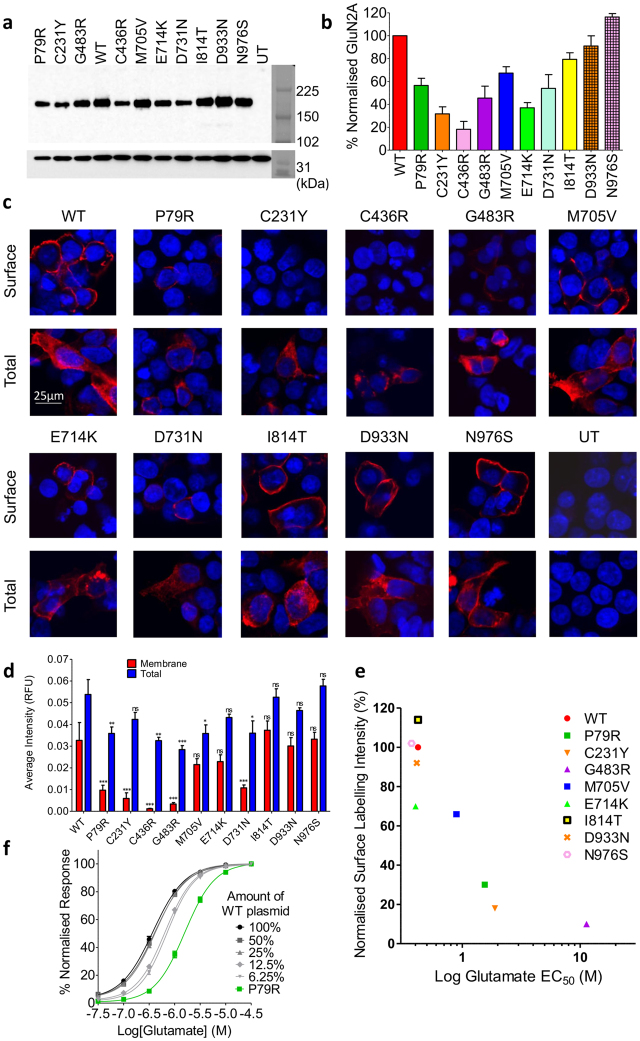
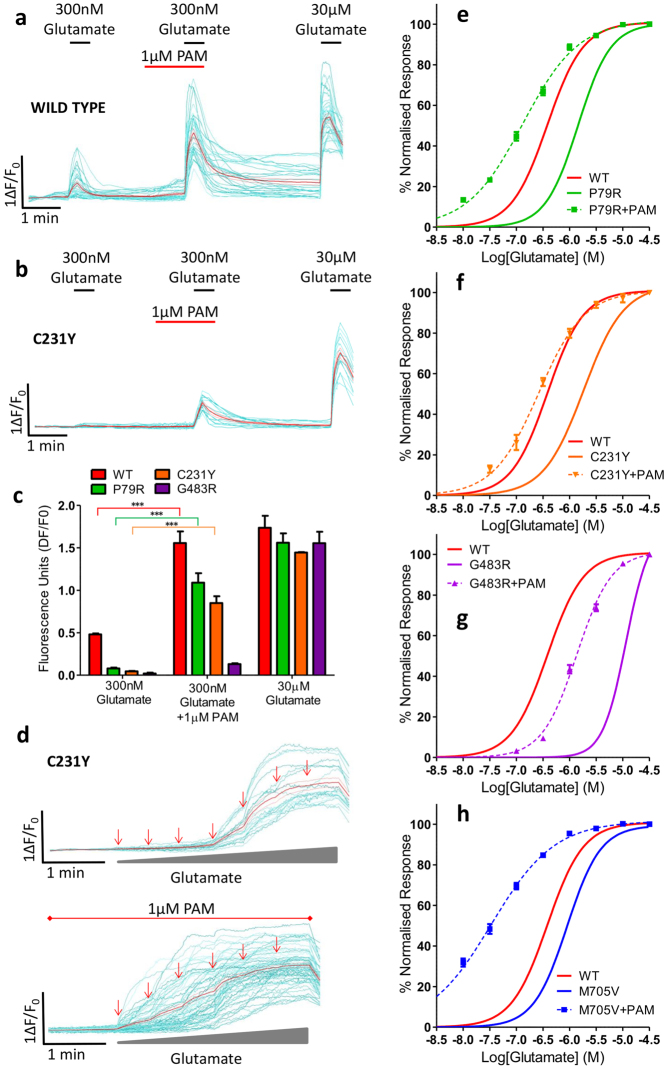
Mutations in the N-methyl-D-aspartate receptor (NMDAR) gene GRIN2A cause epilepsy-aphasia syndrome (EAS), a spectrum of epileptic, cognitive and language disorders. Using bioinformatic and patient data we shortlisted 10 diverse missense mutations for characterisation. We used high-throughput calcium-flux assays and patch clamp recordings of transiently transfected HEK-293 cells for electrophysiological characterization, and Western blotting and confocal imaging to assay expression and surface trafficking. Mutations P79R, C231Y, G483R and M705V caused a significant reduction in glutamate and glycine agonist potency, whilst D731N was non-responsive. These mutants, along with E714K, also showed significantly decreased total protein levels and trafficking to the cell surface, whilst C436R was not trafficked at all. Crucially this reduced surface expression did not cause the reduced agonist response. We were able to rescue the phenotype of P79R, C231Y, G483R and M705V after treatment with a GluN2A-selective positive allosteric modulator. With our methodology we were not able to identify any functional deficits in mutations I814T, D933N and N976S located between the glutamate-binding domain and C-terminus. We show GRIN2A mutations affect the expression and function of the receptor in different ways. Careful molecular profiling of patients will be essential for future effective personalised treatment options.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
