

Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase
- PMID: 28245547
- PMCID: PMC5372010
- DOI: 10.3390/biology6010017
Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase
Abstract
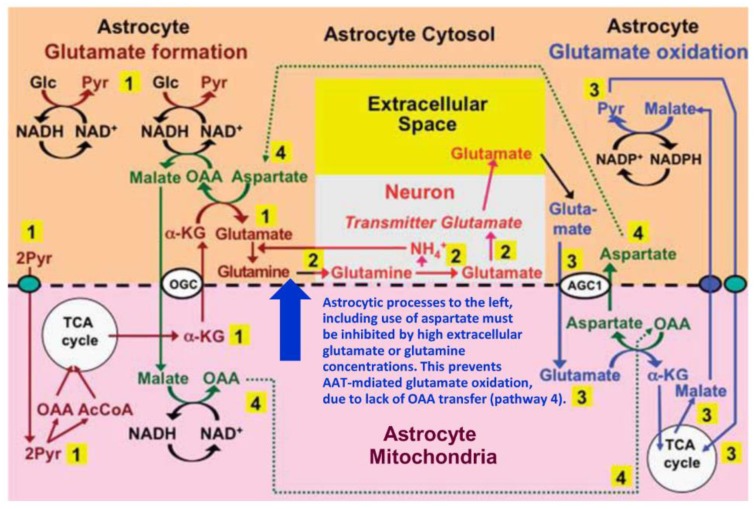
The glutamine-glutamate cycle provides neurons with astrocyte-generated glutamate/γ-aminobutyric acid (GABA) and oxidizes glutamate in astrocytes, and it returns released transmitter glutamate/GABA to neurons after astrocytic uptake. This review deals primarily with the glutamate/GABA generation/oxidation, although it also shows similarity between metabolic rates in cultured astrocytes and intact brain. A key point is identification of the enzyme(s) converting astrocytic α-ketoglutarate to glutamate and vice versa. Most experiments in cultured astrocytes, including those by one of us, suggest that glutamate formation is catalyzed by aspartate aminotransferase (AAT) and its degradation by glutamate dehydrogenase (GDH). Strongly supported by results shown in Table 1 we now propose that both reactions are primarily catalyzed by AAT. This is possible because the formation occurs in the cytosol and the degradation in mitochondria and they are temporally separate. High glutamate/glutamine concentrations abolish the need for glutamate production from α-ketoglutarate and due to metabolic coupling between glutamate synthesis and oxidation these high concentrations render AAT-mediated glutamate oxidation impossible. This necessitates the use of GDH under these conditions, shown by insensitivity of the oxidation to the transamination inhibitor aminooxyacetic acid (AOAA). Experiments using lower glutamate/glutamine concentration show inhibition of glutamate oxidation by AOAA, consistent with the coupled transamination reactions described here.
Keywords: aspartate aminotransferase; astrocyte culture; brain metabolism; glutamate dehydrogenase; glutamate oxidation; glutamate-glutamine cycle; glutamine synthetase; malate-aspartate shuttle; metabolic compartmentation; nitrogen balance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
New insights into the compartmentation of glutamate and glutamine in cultured rat brain astrocytes.Dev Neurosci. 1996;18(5-6):380-90. doi: 10.1159/000111431. Dev Neurosci. 1996. PMID: 8940609
-
Glucose, Lactate, β-Hydroxybutyrate, Acetate, GABA, and Succinate as Substrates for Synthesis of Glutamate and GABA in the Glutamine-Glutamate/GABA Cycle.Adv Neurobiol. 2016;13:9-42. doi: 10.1007/978-3-319-45096-4_2. Adv Neurobiol. 2016. PMID: 27885625
-
Glutamate oxidation in astrocytes: Roles of glutamate dehydrogenase and aminotransferases.J Neurosci Res. 2016 Dec;94(12):1561-1571. doi: 10.1002/jnr.23908. Epub 2016 Sep 15. J Neurosci Res. 2016. PMID: 27629247 Review.
-
Regulation of energy metabolism in synaptic terminals and cultured rat brain astrocytes: differences revealed using aminooxyacetate.Dev Neurosci. 1993;15(3-5):320-9. doi: 10.1159/000111351. Dev Neurosci. 1993. PMID: 7805585
-
The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis.Neurochem Res. 2012 Nov;37(11):2439-55. doi: 10.1007/s11064-012-0803-4. Epub 2012 May 23. Neurochem Res. 2012. PMID: 22618691 Free PMC article. Review.
Cited by
-
Neuropathic Pain and Spinal Cord Injury: Management, Phenotypes, and Biomarkers.Drugs. 2023 Jul;83(11):1001-1025. doi: 10.1007/s40265-023-01903-7. Epub 2023 Jun 16. Drugs. 2023. PMID: 37326804 Review.
-
A Bird's-Eye View of Glycolytic Upregulation in Activated Brain: The Major Fate of Lactate Is Release From Activated Tissue, Not Shuttling to Nearby Neurons.J Neurochem. 2025 Jun;169(6):e70111. doi: 10.1111/jnc.70111. J Neurochem. 2025. PMID: 40476345 Free PMC article. Review.
-
Brain energy metabolism: A roadmap for future research.J Neurochem. 2024 May;168(5):910-954. doi: 10.1111/jnc.16032. Epub 2024 Jan 6. J Neurochem. 2024. PMID: 38183680 Free PMC article. Review.
-
Chronic Valproic Acid Administration Increases Plasma, Liver, and Brain Ammonia Concentration and Suppresses Glutamine Synthetase Activity.Brain Sci. 2020 Oct 21;10(10):759. doi: 10.3390/brainsci10100759. Brain Sci. 2020. PMID: 33096612 Free PMC article.
-
Traumatic brain injury induces region-specific glutamate metabolism changes as measured by multiple mass spectrometry methods.iScience. 2021 Sep 9;24(10):103108. doi: 10.1016/j.isci.2021.103108. eCollection 2021 Oct 22. iScience. 2021. PMID: 34622161 Free PMC article.
References
-
- Smith Q.R. Transport of glutamate and other amino acids at the blood-brain barrier. J. Nutr. 2000;130:1016S–1022S. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
