

Target acquired: Selective autophagy in cardiometabolic disease
- PMID: 28246200
- PMCID: PMC5451512
- DOI: 10.1126/scisignal.aag2298
Target acquired: Selective autophagy in cardiometabolic disease
Abstract
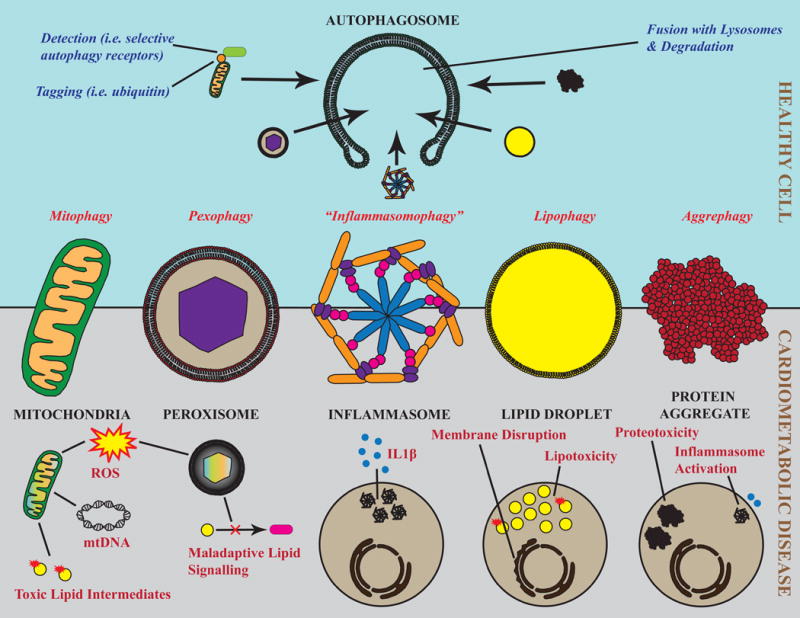
The accumulation of damaged or excess proteins and organelles is a defining feature of metabolic disease in nearly every tissue. Thus, a central challenge in maintaining metabolic homeostasis is the identification, sequestration, and degradation of these cellular components, including protein aggregates, mitochondria, peroxisomes, inflammasomes, and lipid droplets. A primary route through which this challenge is met is selective autophagy, the targeting of specific cellular cargo for autophagic compartmentalization and lysosomal degradation. In addition to its roles in degradation, selective autophagy is emerging as an integral component of inflammatory and metabolic signaling cascades. In this Review, we focus on emerging evidence and key questions about the role of selective autophagy in the cell biology and pathophysiology of metabolic diseases such as obesity, diabetes, atherosclerosis, and steatohepatitis. Essential players in these processes are the selective autophagy receptors, defined broadly as adapter proteins that both recognize cargo and target it to the autophagosome. Additional domains within these receptors may allow integration of information about autophagic flux with critical regulators of cellular metabolism and inflammation. Details regarding the precise receptors involved, such as p62 and NBR1, and their predominant interacting partners are just beginning to be defined. Overall, we anticipate that the continued study of selective autophagy will prove to be informative in understanding the pathogenesis of metabolic diseases and to provide previously unrecognized therapeutic targets.
Copyright © 2017, American Association for the Advancement of Science.
Figures





References
-
- Roth GA, et al. Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation. 2015;132:1667–1678. - PubMed
-
- Patterson C, Ike C, Willis PW, IV, Stouffer GA, Willis MS. The bitter end: The ubiquitin-proteasome system and cardiac dysfunction. Circulation. 2007;115:1456–1463. - PubMed
-
- Versari D, et al. Dysregulation of the ubiquitin-proteasome system in human carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2132–2139. - PubMed
-
- Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147:728–741. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
