

A rare subclinical or mild type of Becker muscular dystrophy caused by a single exon 48 deletion of the dystrophin gene
- PMID: 28247318
- PMCID: PMC5509810
- DOI: 10.1007/s13353-017-0391-8
A rare subclinical or mild type of Becker muscular dystrophy caused by a single exon 48 deletion of the dystrophin gene
Abstract
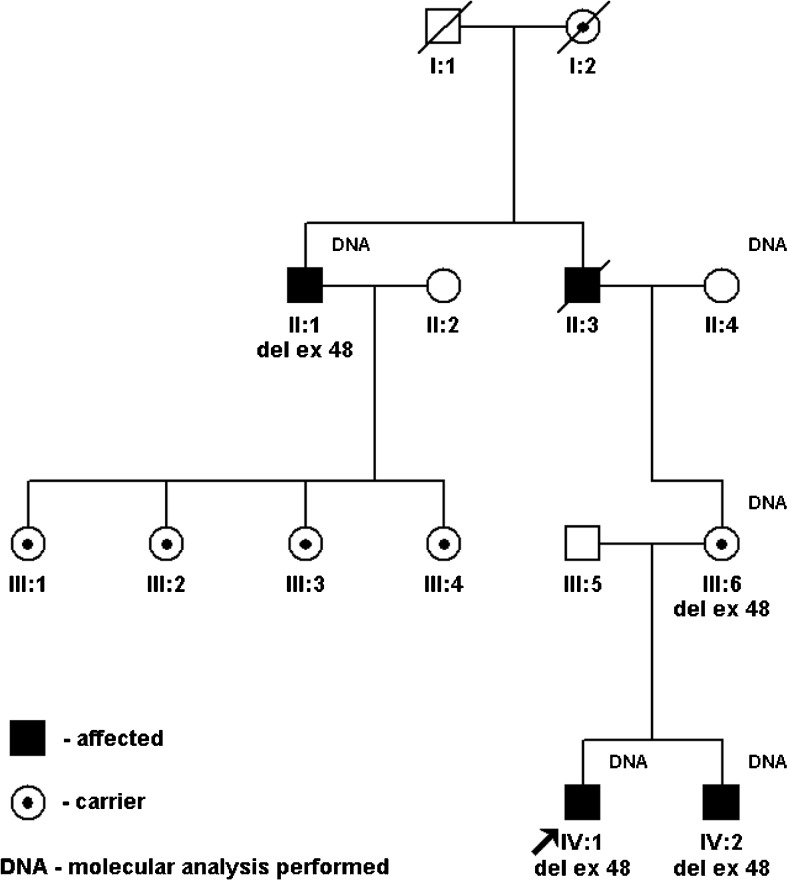
In the material of 227 families with Becker muscular dystrophy (BMD), we found nine non-consanguineous families with 17 male individuals carrying a rare mutation-a single exon 48 deletion of the dystrophin gene-who were affected with a very mild or subclinical form of BMD. They were usually detected thanks to accidental findings of elevated serum creatine phosphokinase (sCPK). A thorough clinical analysis of the carriers, both children (12) and adults (5), revealed in some of them muscle hypotonia (10/17) and/or very mild muscle weakness (9/17), as well as decreased tendon reflexes (6/17). Adults, apart from very mild muscle weakness and calf hypertrophy in some, had no significant abnormalities on neurological assessments and had good exercise tolerance. Parents of the children carriers of the exon 48 deletion are usually unaware of their children being affected, and possibly at risk of developing life-threatening cardiomyopathy. The same concerns the adult male carriers. Therefore, the authors postulate undertaking preventive measures such as cascade screening of the relatives of the probands. Newborn screening programmes of Duchenne muscular dystrophy (DMD)/BMD based on sCPK marked increase may be considered.
Keywords: Asymptomatic mutations; Dystrophin gene; Dystrophinopathy; Exon 48 deletion; Subclinical BMD.
Conflict of interest statement
There is no conflict of interest related to the manuscript on the part of the authors.
Figures
References
-
- Bastianutto C, Bestard JA, Lahnakoski K, Broere D, De Visser M, Zaccolo M, Pozzan T, Ferlini A, Muntoni F, Patarnello T, Klamut HJ. Dystrophin muscle enhancer 1 is implicated in the activation of non-muscle isoforms in the skeletal muscle of patients with X-linked dilated cardiomyopathy. Hum Mol Genet. 2001;10:2627–2635. doi: 10.1093/hmg/10.23.2627. - DOI - PubMed
-
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595–605. doi: 10.1016/S0140-6736(11)60756-3. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
