

Mechanisms of Immune Signaling in Colitis-Associated Cancer
- PMID: 28247866
- PMCID: PMC5301162
- DOI: 10.1016/j.jcmgh.2014.11.006
Mechanisms of Immune Signaling in Colitis-Associated Cancer
Abstract
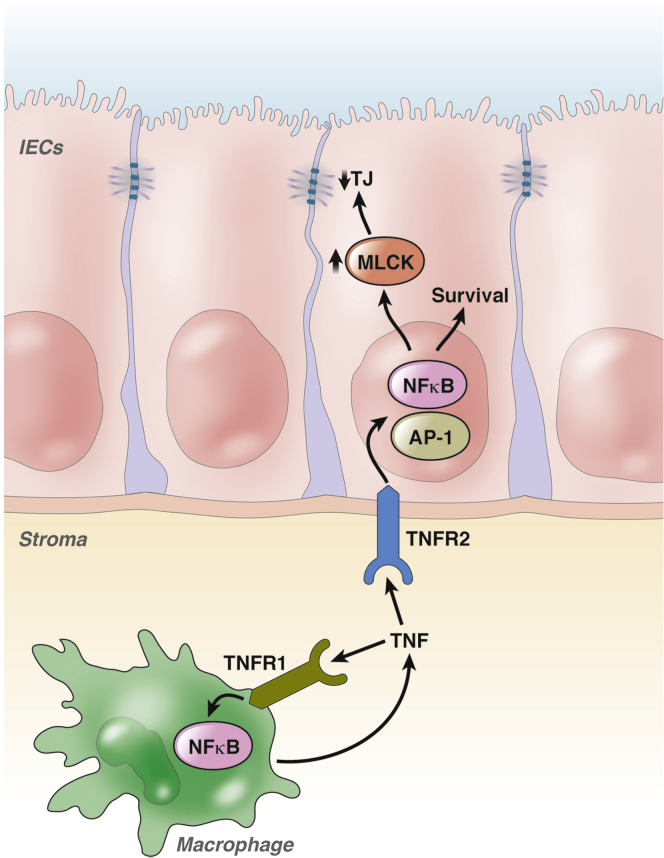
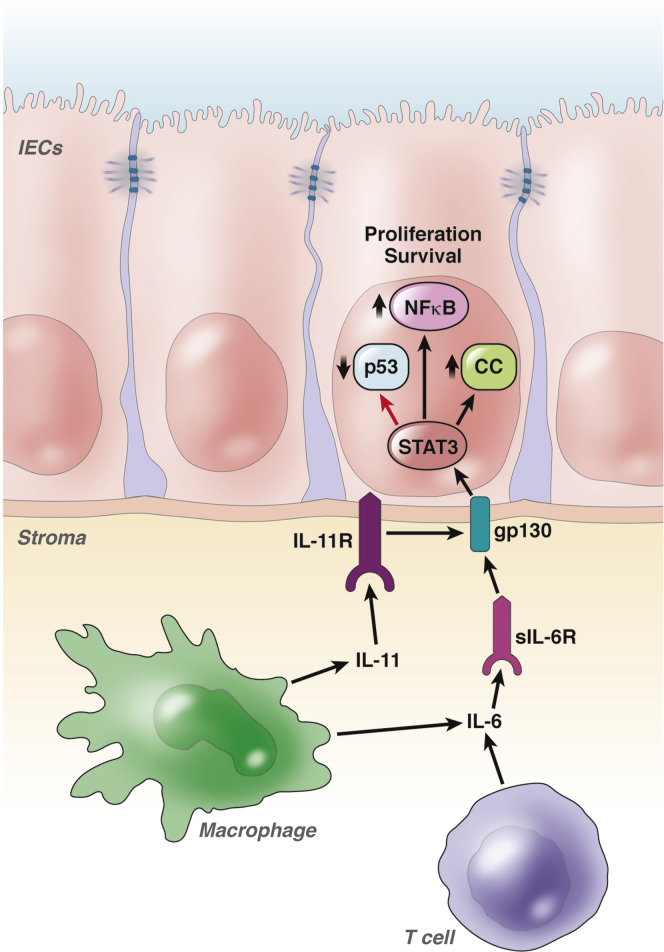
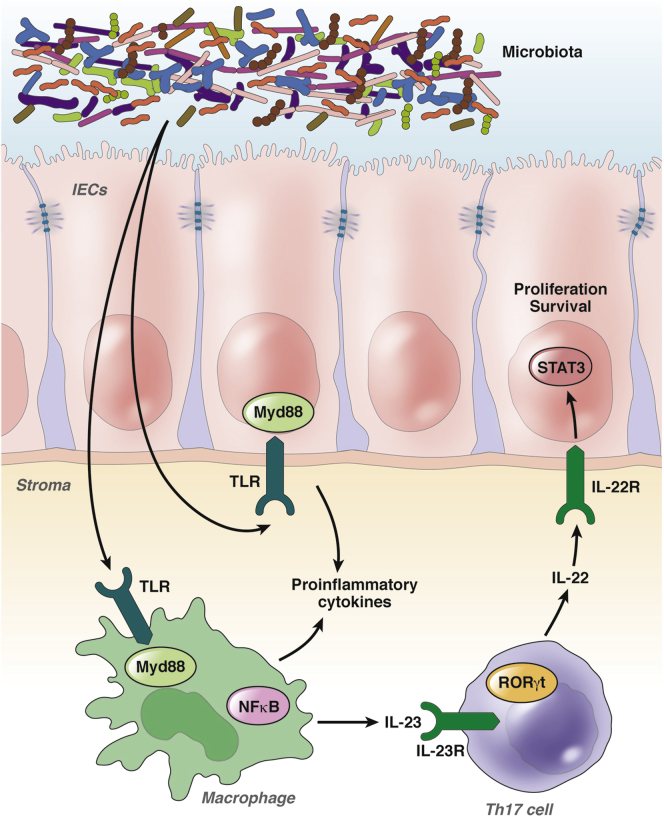
The inflammatory bowel diseases ulcerative colitis and Crohn's disease are associated with an increased risk for the development of colorectal cancer. During recent years, several immune signaling pathways have been linked to colitis-associated cancer (CAC), largely owing to the availability of suitable preclinical models. Among these, chronic intestinal inflammation has been shown to support tumor initiation through oxidative stress-induced mutations. A proinflammatory microenvironment that develops, possibly as a result of defective intestinal barrier function and host-microbial interactions, enables tumor promotion. Several molecular pathways such as tumor necrosis factor/nuclear factor-κB or interleukin 6/signal transducer and activator of transcription 3 signaling have been identified as important contributors to CAC development and could be promising therapeutic targets for the prevention and treatment of CAC.
Keywords: AOM-DSS, azoxymethane–dextran sulfate sodium; APC, adenomatous polyposis coli; CAC, colitis-associated cancer; CD, Crohn’s disease; CRC, colorectal cancer; Colorectal Cancer; Crohn's Disease; Cytokines; DDR, DNA damage response; IBD, inflammatory bowel disease; IKK, IκB kinase; IL, interleukin; IL6R, interleukin 6 receptor; Inflammatory Bowel Disease; Interleukin-6; LPS, lipopolysaccharide; Myd88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor-κB; NLR, NOD- and leucine-rich repeat–containing protein; NLRP, nucleotide-binding oligomerization domain- and leucine-rich repeat–containing protein family, pyrin domain-containing; NOD, nucleotide-binding oligomerization domain; RONS, reactive oxygen and nitrogen species; STAT3, signal transducer and activator of transcription 3; TLR, Toll-like receptor; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; Th17, T-helper 17; Tumor Necrosis Factor Alpha; UC, ulcerative colitis; Ulcerative Colitis; gp, glycoprotein.
Figures
References
-
- Balkwill F., Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. - PubMed
-
- Terzic J., Grivennikov S., Karin E. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–2114 e5. - PubMed
-
- Crohn B., Rosenberg H. The sigmoidoscopic picture of chronic ulcerative colitis (non-specific) Am J Med Sci. 1925;170:220–228.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
