

Water Dynamics in the Hydration Shells of Biomolecules
- PMID: 28248491
- PMCID: PMC5571470
- DOI: 10.1021/acs.chemrev.6b00765
Water Dynamics in the Hydration Shells of Biomolecules
Abstract
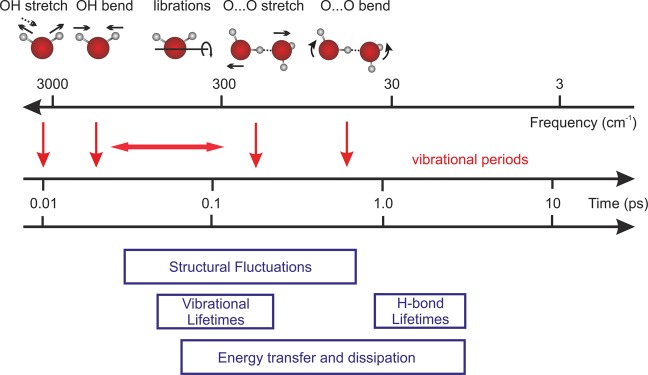
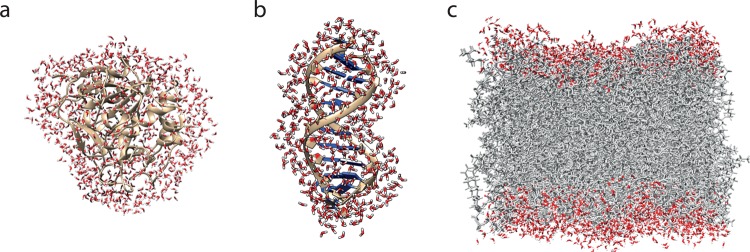
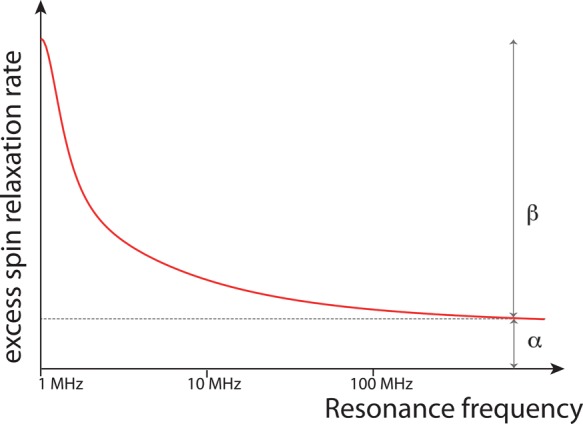
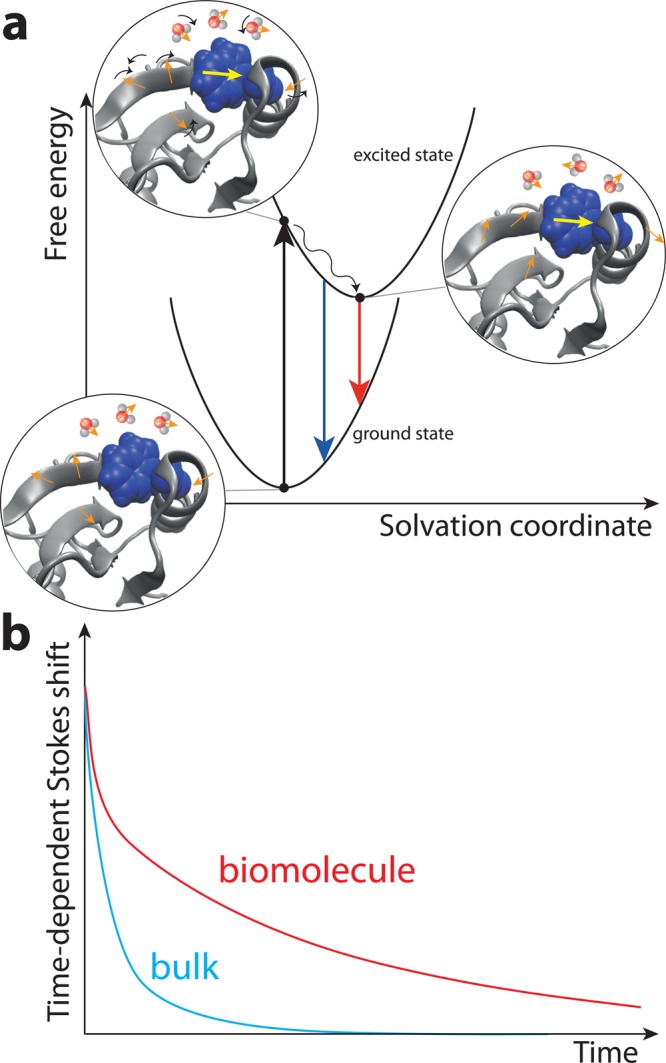
The structure and function of biomolecules are strongly influenced by their hydration shells. Structural fluctuations and molecular excitations of hydrating water molecules cover a broad range in space and time, from individual water molecules to larger pools and from femtosecond to microsecond time scales. Recent progress in theory and molecular dynamics simulations as well as in ultrafast vibrational spectroscopy has led to new and detailed insight into fluctuations of water structure, elementary water motions, electric fields at hydrated biointerfaces, and processes of vibrational relaxation and energy dissipation. Here, we review recent advances in both theory and experiment, focusing on hydrated DNA, proteins, and phospholipids, and compare dynamics in the hydration shells to bulk water.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
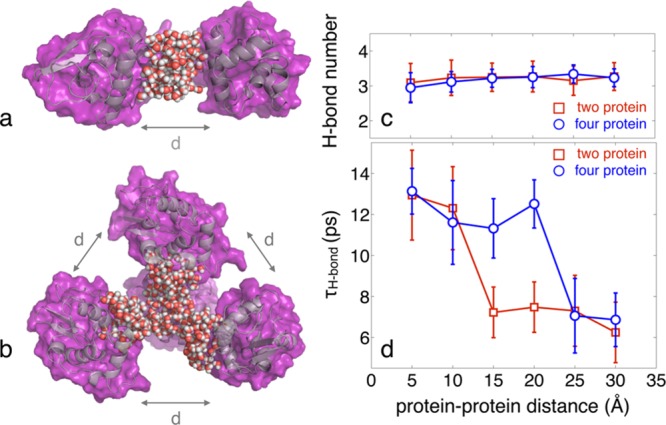
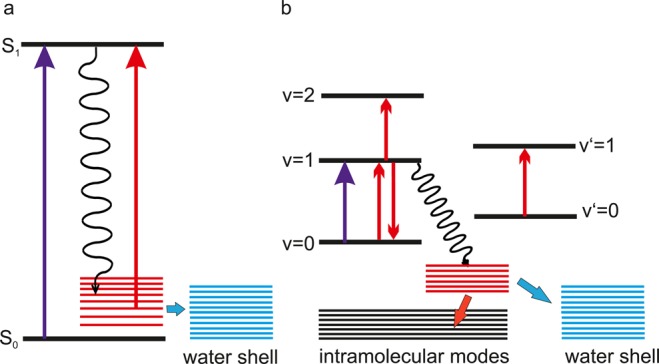
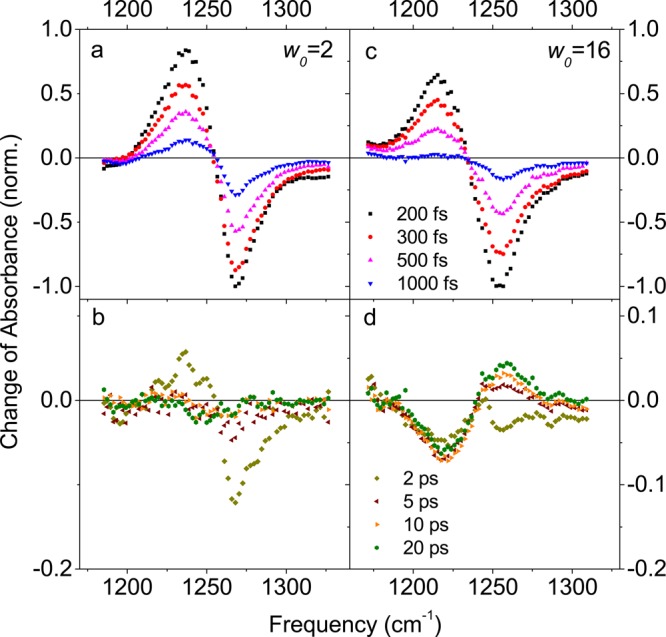
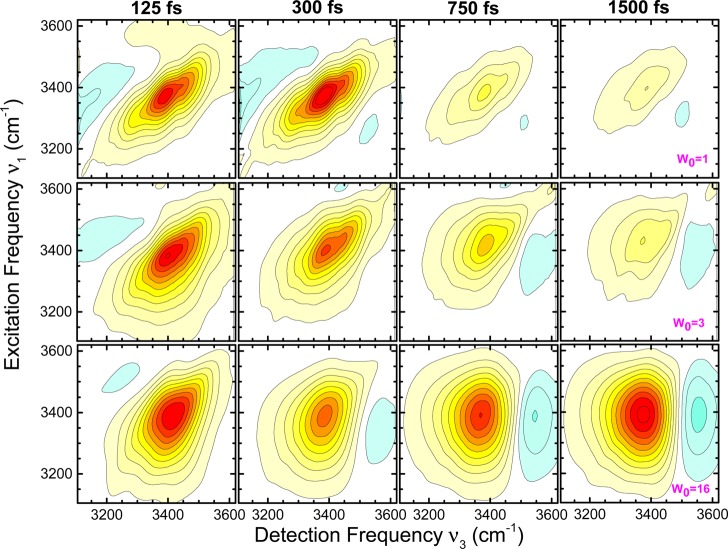
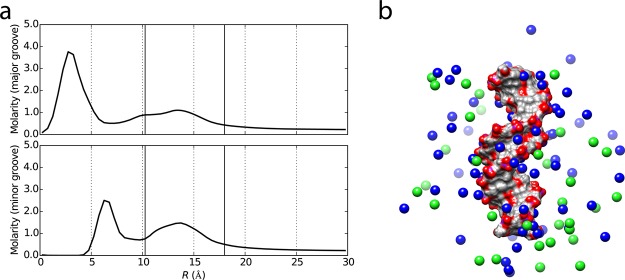
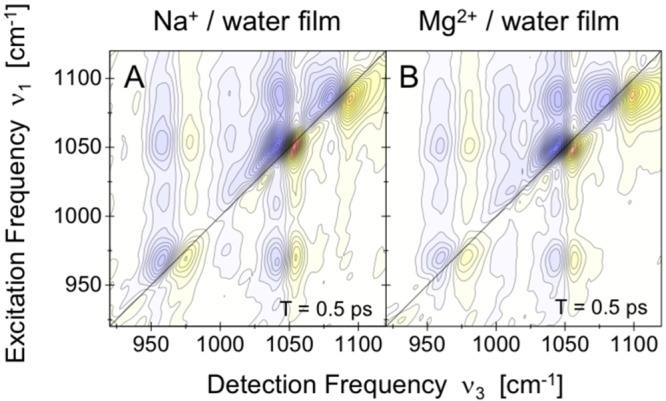
References
-
- Arunan E.; Desiraju G. R.; Klein R. A.; Sadlej J.; Scheiner S.; Alkorta I.; Clary D. C.; Crabtree R. H.; Dannenberg J. J.; Hobza P.; et al. Defining the Hydrogen Bond: An Account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. 10.1351/PAC-REP-10-01-01. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
