

Biological determinants of radioresistance and their remediation in pancreatic cancer
- PMID: 28249796
- PMCID: PMC5548591
- DOI: 10.1016/j.bbcan.2017.02.003
Biological determinants of radioresistance and their remediation in pancreatic cancer
Abstract
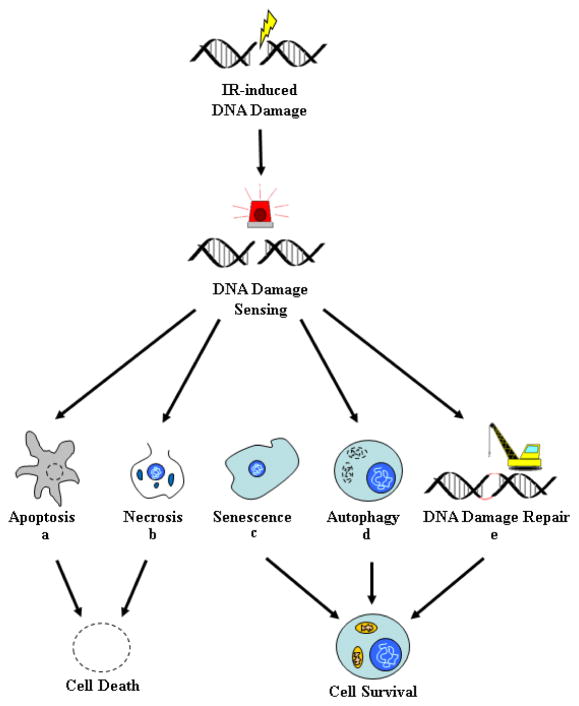
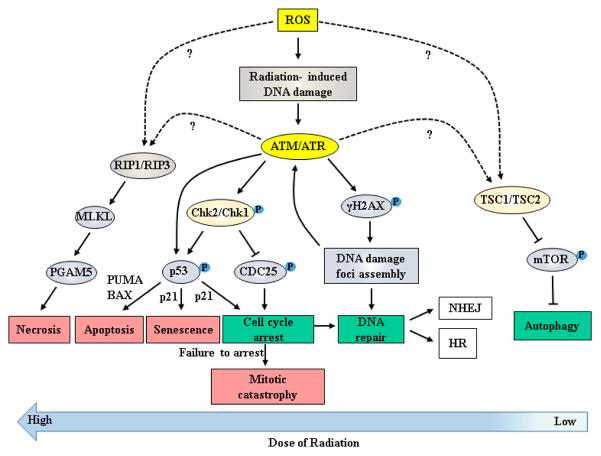
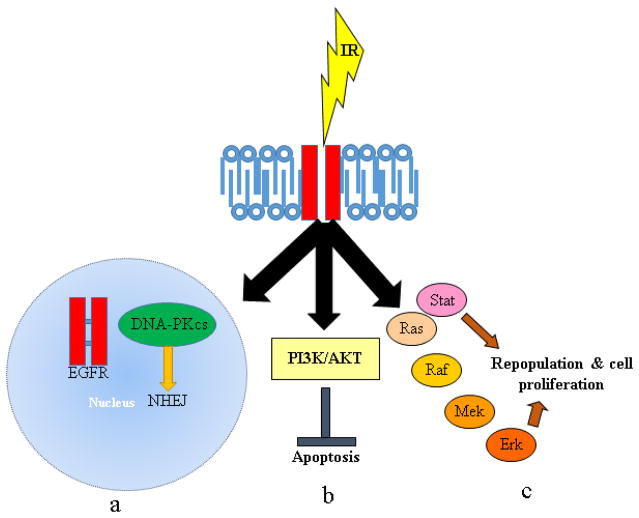
Despite recent advances in radiotherapy, a majority of patients diagnosed with pancreatic cancer (PC) do not achieve objective responses due to the existence of intrinsic and acquired radioresistance. Identification of molecular mechanisms that compromise the efficacy of radiation therapy and targeting these pathways is paramount for improving radiation response in PC patients. In this review, we have summarized molecular mechanisms associated with the radio-resistant phenotype of PC. Briefly, we discuss the reversible and irreversible biological consequences of radiotherapy, such as DNA damage and DNA repair, mechanisms of cancer cell survival and radiation-induced apoptosis following radiotherapy. We further describe various small molecule inhibitors and molecular targeting agents currently being tested in preclinical and clinical studies as potential radiosensitizers for PC. Notably, we draw attention towards the confounding effects of cancer stem cells, immune system, and the tumor microenvironment in the context of PC radioresistance and radiosensitization. Finally, we discuss the need for examining selective radioprotectors in light of the emerging evidence on radiation toxicity to non-target tissue associated with PC radiotherapy.
Copyright © 2017 Elsevier B.V. All rights reserved.
Conflict of interest statement
None
Figures
References
-
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. - PubMed
-
- National Cancer Institute. Surveillance Epidemiology and End Results Cancer Statistics Review 1975–2006. 2011
-
- Barugola G, Falconi M, Bettini R, Boninsegna L, Casarotto A, Salvia R, Bassi C, Pederzoli P. The determinant factors of recurrence following resection for ductal pancreatic cancer. JOP. 2007;8:132–140. - PubMed
-
- Barugola G, Partelli S, Marcucci S, Sartori N, Capelli P, Bassi C, Pederzoli P, Falconi M. Resectable pancreatic cancer: who really benefits from resection? Ann Surg Oncol. 2009;16:3316–3322. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
