

Inference of the Distribution of Selection Coefficients for New Nonsynonymous Mutations Using Large Samples
- PMID: 28249985
- PMCID: PMC5419480
- DOI: 10.1534/genetics.116.197145
Inference of the Distribution of Selection Coefficients for New Nonsynonymous Mutations Using Large Samples
Abstract
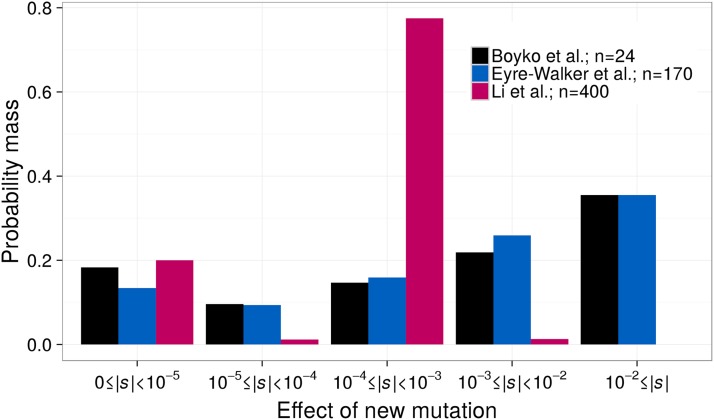
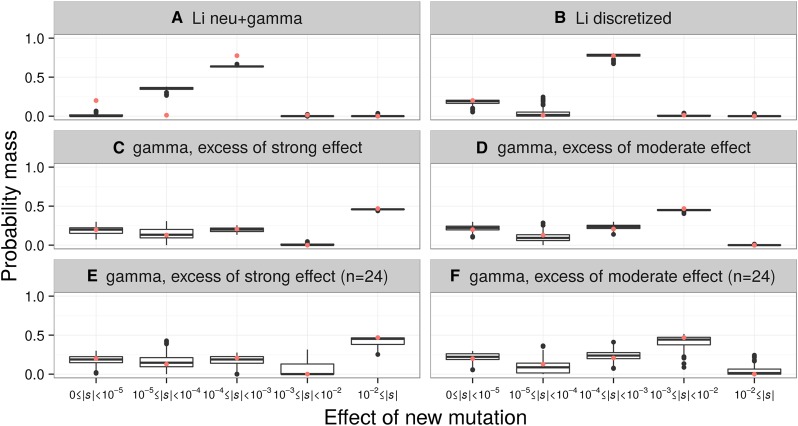
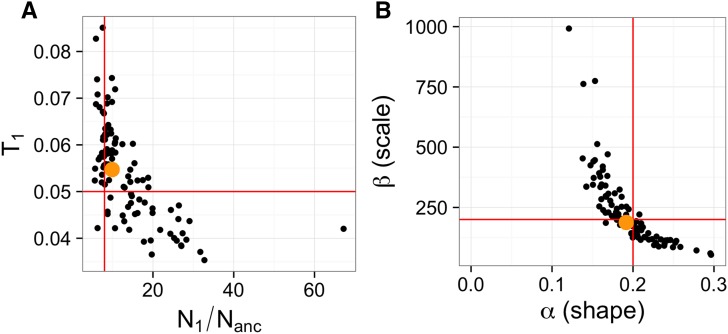
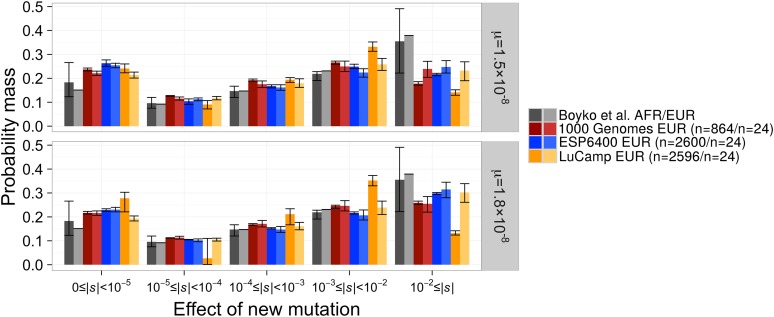
The distribution of fitness effects (DFE) has considerable importance in population genetics. To date, estimates of the DFE come from studies using a small number of individuals. Thus, estimates of the proportion of moderately to strongly deleterious new mutations may be unreliable because such variants are unlikely to be segregating in the data. Additionally, the true functional form of the DFE is unknown, and estimates of the DFE differ significantly between studies. Here we present a flexible and computationally tractable method, called Fit∂a∂i, to estimate the DFE of new mutations using the site frequency spectrum from a large number of individuals. We apply our approach to the frequency spectrum of 1300 Europeans from the Exome Sequencing Project ESP6400 data set, 1298 Danes from the LuCamp data set, and 432 Europeans from the 1000 Genomes Project to estimate the DFE of deleterious nonsynonymous mutations. We infer significantly fewer (0.38-0.84 fold) strongly deleterious mutations with selection coefficient |s| > 0.01 and more (1.24-1.43 fold) weakly deleterious mutations with selection coefficient |s| < 0.001 compared to previous estimates. Furthermore, a DFE that is a mixture distribution of a point mass at neutrality plus a gamma distribution fits better than a gamma distribution in two of the three data sets. Our results suggest that nearly neutral forces play a larger role in human evolution than previously thought.
Keywords: deleterious mutations; diffusion theory; population genetics; site frequency spectrum.
Copyright © 2017 by the Genetics Society of America.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
