

Systemic autoimmunity induced by the TLR7/8 agonist Resiquimod causes myocarditis and dilated cardiomyopathy in a new mouse model of autoimmune heart disease
- PMID: 28250051
- PMCID: PMC5374321
- DOI: 10.1242/dmm.027409
Systemic autoimmunity induced by the TLR7/8 agonist Resiquimod causes myocarditis and dilated cardiomyopathy in a new mouse model of autoimmune heart disease
Abstract
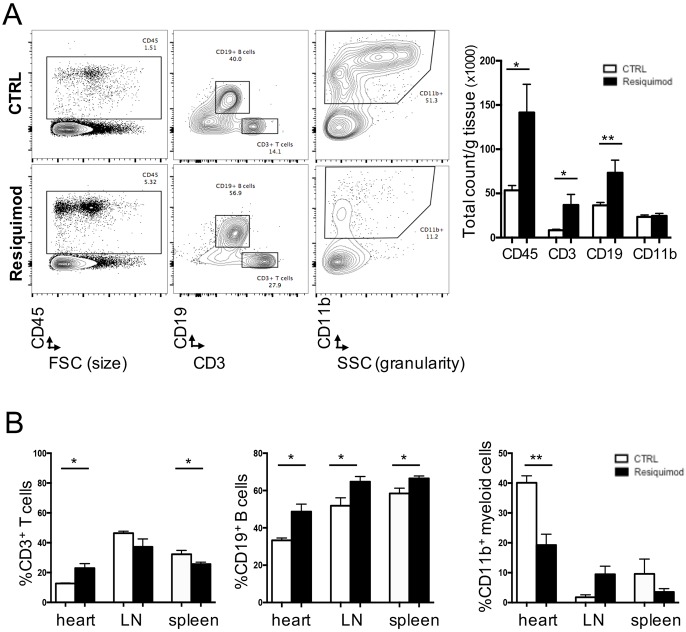
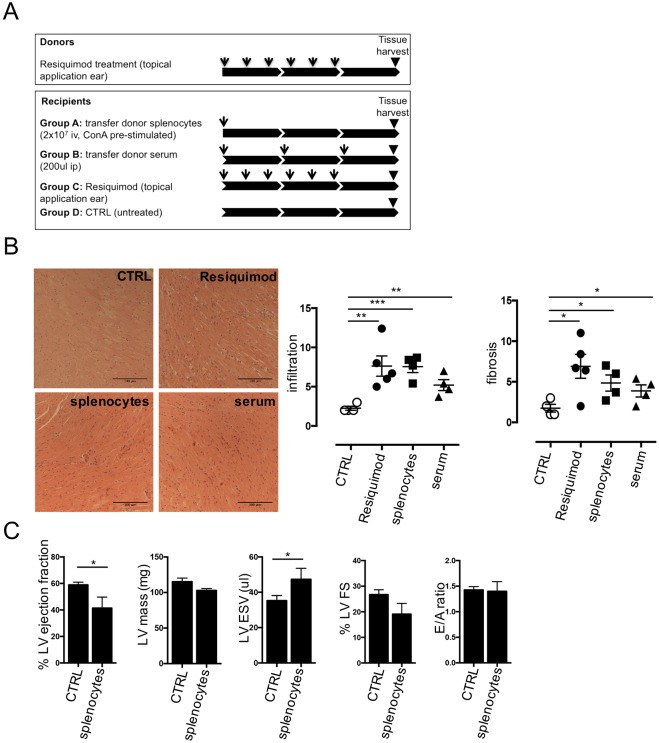
Systemic autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) show significant heart involvement and cardiovascular morbidity, which can be due to systemically increased levels of inflammation or direct autoreactivity targeting cardiac tissue. Despite high clinical relevance, cardiac damage secondary to systemic autoimmunity lacks inducible rodent models. Here, we characterise immune-mediated cardiac tissue damage in a new model of SLE induced by topical application of the Toll-like receptor 7/8 (TLR7/8) agonist Resiquimod. We observe a cardiac phenotype reminiscent of autoimmune-mediated dilated cardiomyopathy, and identify auto-antibodies as major contributors to cardiac tissue damage. Resiquimod-induced heart disease is a highly relevant mouse model for mechanistic and therapeutic studies aiming to protect the heart during autoimmunity.
Keywords: Autoimmunity; Dilated cardiomyopathy; Heart disease; Model; Myocarditis; Resiquimod; Toll-like receptor 7/8.
© 2017. Published by The Company of Biologists Ltd.
Conflict of interest statement
The authors declare no competing or financial interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
