

Biallelic mutations in human DCC cause developmental split-brain syndrome
- PMID: 28250456
- PMCID: PMC5374027
- DOI: 10.1038/ng.3804
Biallelic mutations in human DCC cause developmental split-brain syndrome
Abstract
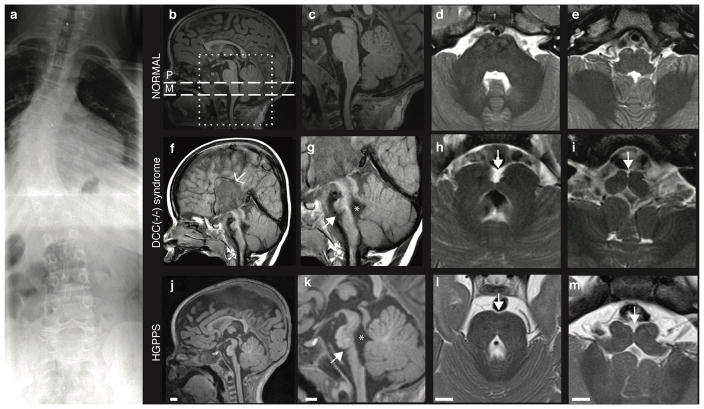
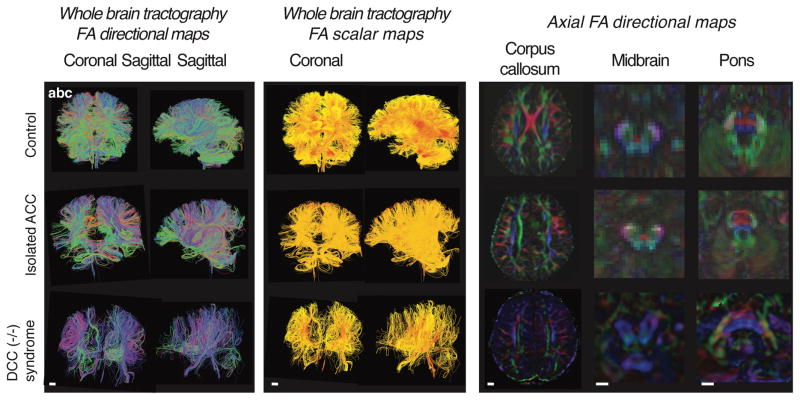
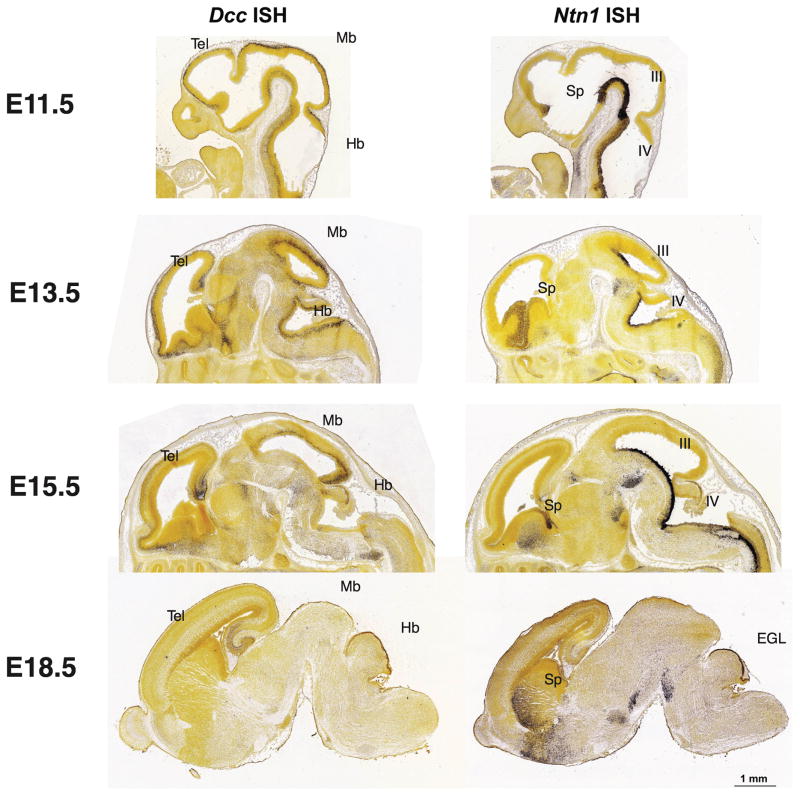
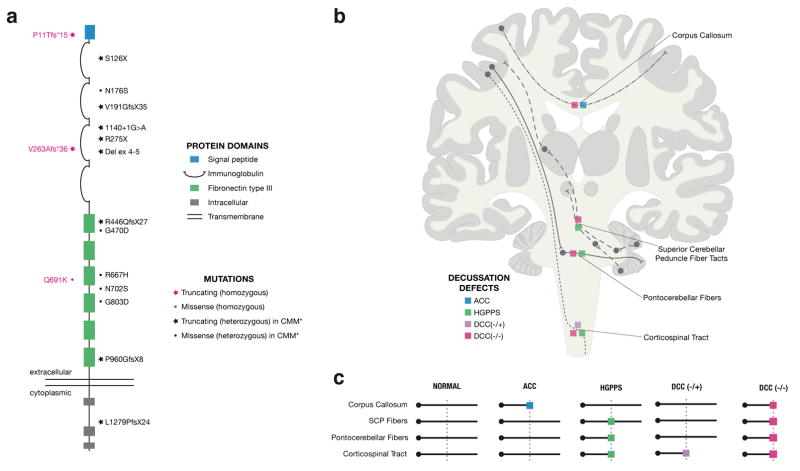
Motor, sensory, and integrative activities of the brain are coordinated by a series of midline-bridging neuronal commissures whose development is tightly regulated. Here we report a new human syndrome in which these commissures are widely disrupted, thus causing clinical manifestations of horizontal gaze palsy, scoliosis, and intellectual disability. Affected individuals were found to possess biallelic loss-of-function mutations in the gene encoding the axon-guidance receptor 'deleted in colorectal carcinoma' (DCC), which has been implicated in congenital mirror movements when it is mutated in the heterozygous state but whose biallelic loss-of-function human phenotype has not been reported. Structural MRI and diffusion tractography demonstrated broad disorganization of white-matter tracts throughout the human central nervous system (CNS), including loss of all commissural tracts at multiple levels of the neuraxis. Combined with data from animal models, these findings show that DCC is a master regulator of midline crossing and development of white-matter projections throughout the human CNS.
Conflict of interest statement
The authors disclose that T.W.Y. is co-founder of Claritas Genomics, a gene diagnostic and genomic medicine company, and S.S.J. is co-founder of Global Gene Corporation Pte Ltd, a gene diagnostic company; however, T.W.Y. and S.S.J. declare no competing financial interests related to the publication of this work. The remaining authors do not have any financial conflict of interest that might be construed to influence the results or interpretation of this work.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
