

Clinical Features and Genetic Analysis of 48 Patients with Chronic Granulomatous Disease in a Single Center Study from Shanghai, China (2005-2015): New Studies and a Literature Review
- PMID: 28251166
- PMCID: PMC5303869
- DOI: 10.1155/2017/8745254
Clinical Features and Genetic Analysis of 48 Patients with Chronic Granulomatous Disease in a Single Center Study from Shanghai, China (2005-2015): New Studies and a Literature Review
Abstract
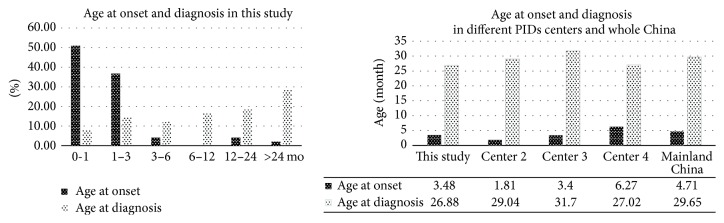
Chronic Granulomatous Disease (CGD) is a rare inherited primary immunodeficiency, which is characterized by recurrent infections due to defective phagocyte NADPH oxidase enzyme. Nowadays, little is known about Chinese CGD patients. Here we report 48 CGD patients in our single center study, which is the largest cohort study from Mainland China. The ratio of male to female was 11 : 1. The mean onset age was 0.29 years old, and 52% patients had an onset within the 1st month of life. The mean diagnosis age was 2.24 years old. 11 patients (23%) had died with an average age of 2.91 years old. 13 patients (28%) had positive family histories. The most prevalent infectious sites were the lungs (77%), followed by gastrointestinal tract (54%), lymph nodes (50%), and skin (46%). In addition, septicopyemia, thrush, and hepatosplenomegaly were also commonly observed, accounting for 23%, 23%, and 40% of the cases. Lesions due to BCG vaccination occurred in more than half of the patients. X-linked CGD due to CYBB gene mutations accounted for 75% of the cases, and 11 of them were novel mutations. Autosomal recessive inheritance accounted for 6% patients, including 1 patient with CYBA, 1 with NCF1, and 1 with NCF2 gene mutations.
Conflict of interest statement
The authors declare that there is no conflict of interests regarding the publication of this paper.
Figures
Similar articles
-
Clinical and Genotypic Spectrum of Chronic Granulomatous Disease in 71 Latin American Patients: First Report from the LASID Registry.Pediatr Blood Cancer. 2015 Dec;62(12):2101-7. doi: 10.1002/pbc.25674. Epub 2015 Jul 15. Pediatr Blood Cancer. 2015. PMID: 26185101 Clinical Trial.
-
A Cohort of 169 Chronic Granulomatous Disease Patients Exposed to BCG Vaccination: a Retrospective Study from a Single Center in Shanghai, China (2004-2017).J Clin Immunol. 2018 Apr;38(3):260-272. doi: 10.1007/s10875-018-0486-y. Epub 2018 Mar 20. J Clin Immunol. 2018. PMID: 29560547
-
Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients.J Allergy Clin Immunol. 2013 Nov;132(5):1156-1163.e5. doi: 10.1016/j.jaci.2013.05.039. Epub 2013 Jul 31. J Allergy Clin Immunol. 2013. PMID: 23910690
-
Chronic granulomatous disease: a 25-year patient registry based on a multistep diagnostic procedure, from the referral center for primary immunodeficiencies in Greece.J Clin Immunol. 2013 Nov;33(8):1302-9. doi: 10.1007/s10875-013-9940-z. Epub 2013 Oct 1. J Clin Immunol. 2013. PMID: 24081483 Review.
-
[Chronic-granulomatous disease].Rev Med Interne. 2009 Mar;30(3):221-32. doi: 10.1016/j.revmed.2008.05.023. Epub 2008 Jul 21. Rev Med Interne. 2009. PMID: 18640747 Review. French.
Cited by
-
X-Linked Chronic Granulomatous Disease: Initial Presentation with Intracranial Hemorrhage from Vitamin K Deficiency in Infant.Case Rep Pediatr. 2018 Jun 24;2018:7041204. doi: 10.1155/2018/7041204. eCollection 2018. Case Rep Pediatr. 2018. PMID: 30034904 Free PMC article.
-
Variant Type X91+ Chronic Granulomatous Disease: Clinical and Molecular Characterization in a Chinese Cohort.J Clin Immunol. 2022 Oct;42(7):1564-1579. doi: 10.1007/s10875-022-01324-3. Epub 2022 Jul 7. J Clin Immunol. 2022. PMID: 35796921 Free PMC article.
-
Achromobacter xylosoxidans Pneumonia in a Young Child with Chronic Granulomatous Disease-a Case-Based Review.J Clin Immunol. 2021 Oct;41(7):1686-1692. doi: 10.1007/s10875-021-01079-3. Epub 2021 Jul 14. J Clin Immunol. 2021. PMID: 34263392 Review. No abstract available.
-
Phenomic Analysis of Chronic Granulomatous Disease Reveals More Severe Integumentary Infections in X-Linked Compared With Autosomal Recessive Chronic Granulomatous Disease.Front Immunol. 2022 Jan 24;12:803763. doi: 10.3389/fimmu.2021.803763. eCollection 2021. Front Immunol. 2022. PMID: 35140711 Free PMC article.
-
A novel variant in the neutrophil cytosolic factor 2 (NCF2) gene results in severe disseminated BCG infectious disease: A clinical report and literature review.Mol Genet Genomic Med. 2020 Jun;8(6):e1237. doi: 10.1002/mgg3.1237. Epub 2020 Apr 12. Mol Genet Genomic Med. 2020. PMID: 32281309 Free PMC article. Review.
References
-
- Berendes H., Bridges R. A., Good R. A. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minnesota Medicine. 1957;40(5):309–312. - PubMed
-
- Landing B. H., Shirkey H. S. A syndrome of recurrent infection and infiltration of viscera by pigmented lipid histiocytes. Pediatrics. 1957;20(3):431–438. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
