

Normalization and microbial differential abundance strategies depend upon data characteristics
- PMID: 28253908
- PMCID: PMC5335496
- DOI: 10.1186/s40168-017-0237-y
Normalization and microbial differential abundance strategies depend upon data characteristics
Abstract
Background: Data from 16S ribosomal RNA (rRNA) amplicon sequencing present challenges to ecological and statistical interpretation. In particular, library sizes often vary over several ranges of magnitude, and the data contains many zeros. Although we are typically interested in comparing relative abundance of taxa in the ecosystem of two or more groups, we can only measure the taxon relative abundance in specimens obtained from the ecosystems. Because the comparison of taxon relative abundance in the specimen is not equivalent to the comparison of taxon relative abundance in the ecosystems, this presents a special challenge. Second, because the relative abundance of taxa in the specimen (as well as in the ecosystem) sum to 1, these are compositional data. Because the compositional data are constrained by the simplex (sum to 1) and are not unconstrained in the Euclidean space, many standard methods of analysis are not applicable. Here, we evaluate how these challenges impact the performance of existing normalization methods and differential abundance analyses.
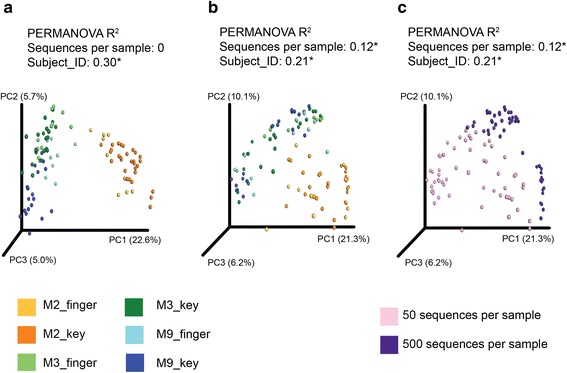
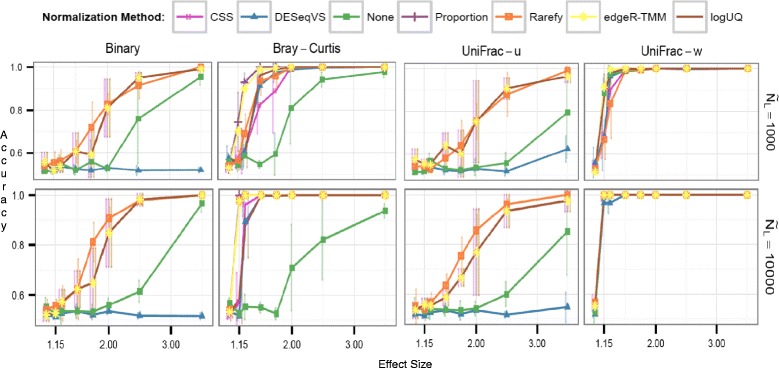
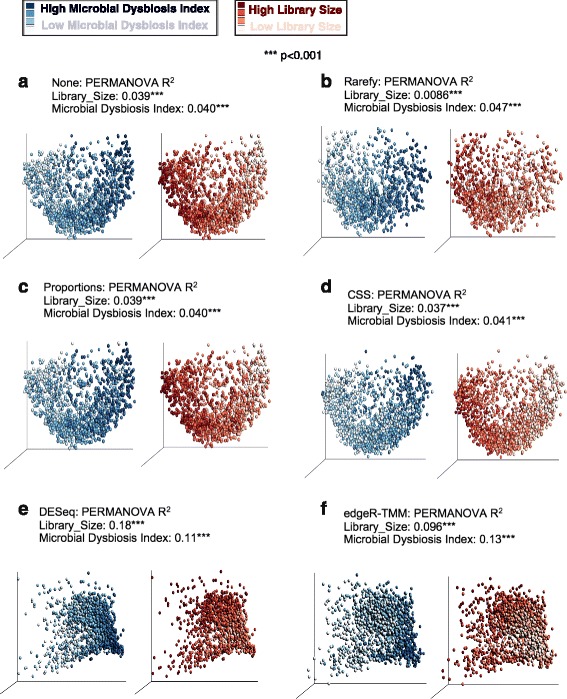
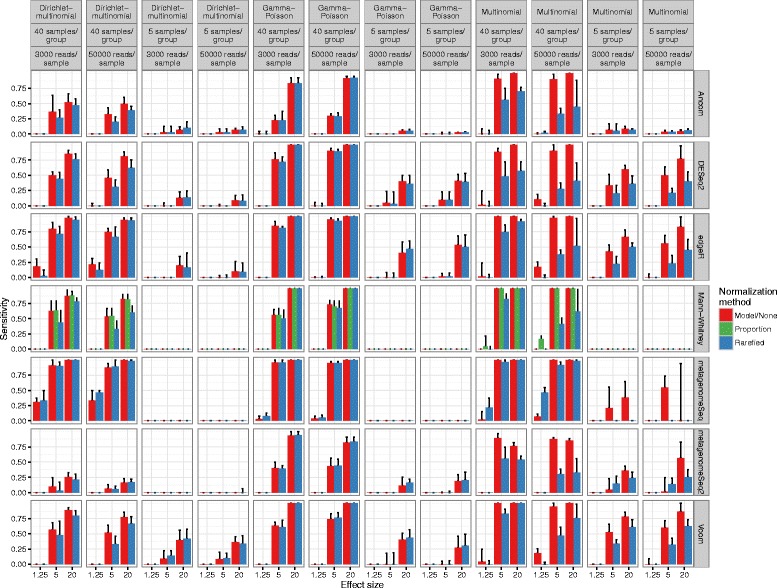
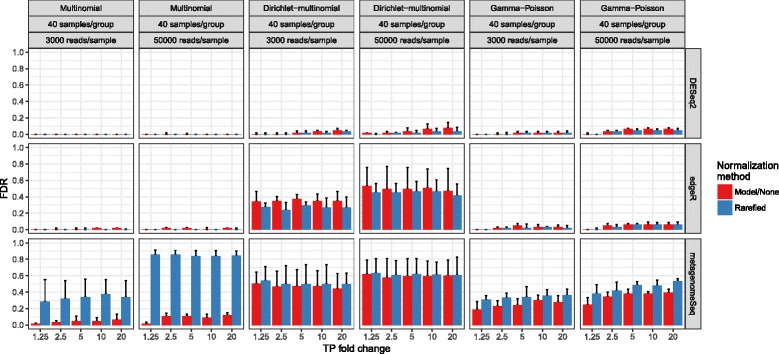
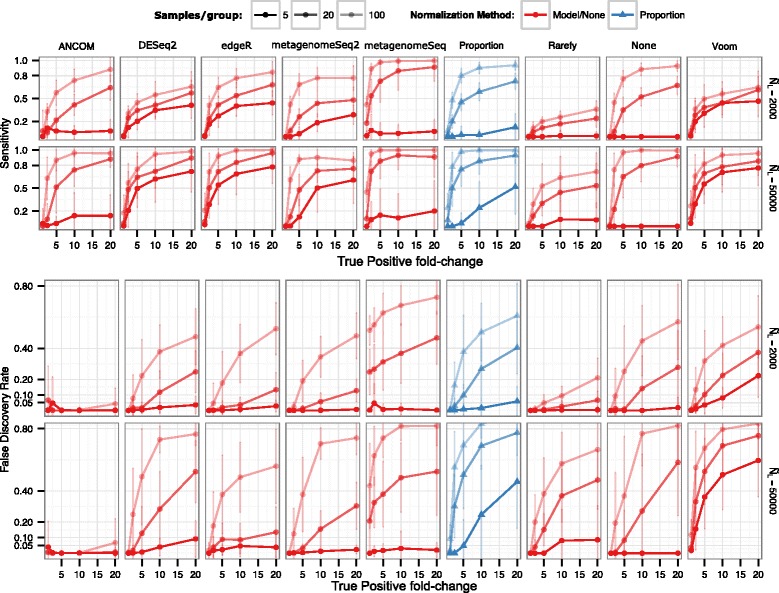
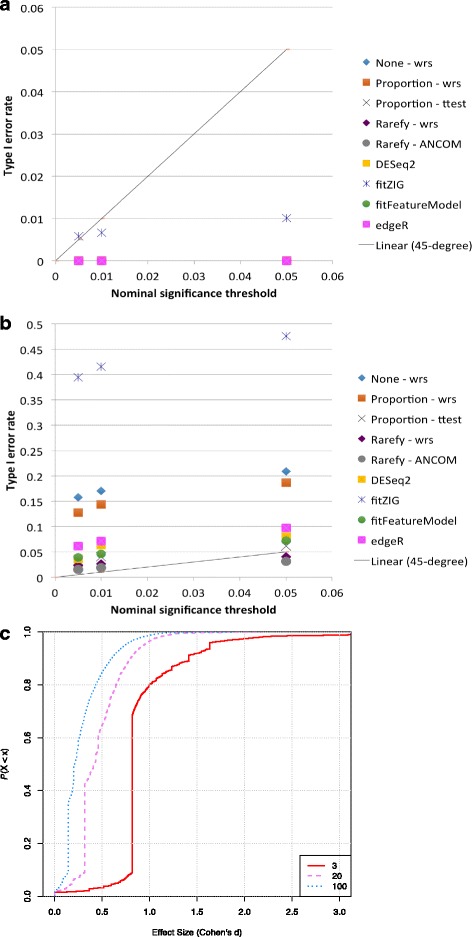
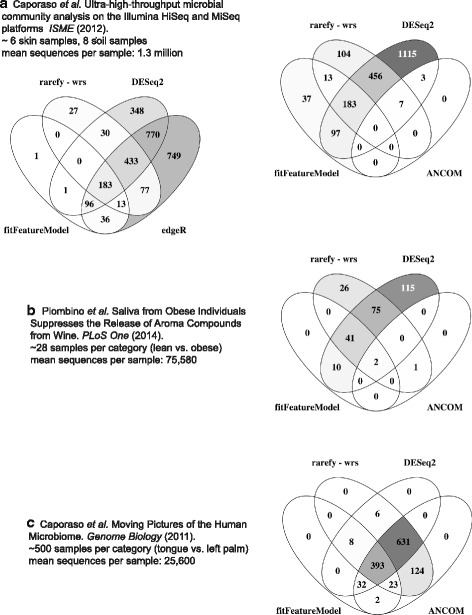
Results: Effects on normalization: Most normalization methods enable successful clustering of samples according to biological origin when the groups differ substantially in their overall microbial composition. Rarefying more clearly clusters samples according to biological origin than other normalization techniques do for ordination metrics based on presence or absence. Alternate normalization measures are potentially vulnerable to artifacts due to library size. Effects on differential abundance testing: We build on a previous work to evaluate seven proposed statistical methods using rarefied as well as raw data. Our simulation studies suggest that the false discovery rates of many differential abundance-testing methods are not increased by rarefying itself, although of course rarefying results in a loss of sensitivity due to elimination of a portion of available data. For groups with large (~10×) differences in the average library size, rarefying lowers the false discovery rate. DESeq2, without addition of a constant, increased sensitivity on smaller datasets (<20 samples per group) but tends towards a higher false discovery rate with more samples, very uneven (~10×) library sizes, and/or compositional effects. For drawing inferences regarding taxon abundance in the ecosystem, analysis of composition of microbiomes (ANCOM) is not only very sensitive (for >20 samples per group) but also critically the only method tested that has a good control of false discovery rate.
Conclusions: These findings guide which normalization and differential abundance techniques to use based on the data characteristics of a given study.
Keywords: Differential abundance; Microbiome; Normalization; Statistics.
Figures
References
-
- Aitchison J. The statistical analysis of compositional data. J Roy Stat Soc B Met. 1982;44(2):139–177.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
