

Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer
- PMID: 28255083
- PMCID: PMC5380483
- DOI: 10.1158/2159-8290.CD-16-0611
Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer
Erratum in
-
Correction: Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer.Cancer Discov. 2017 Jul;7(7):782. doi: 10.1158/2159-8290.CD-17-0565. Epub 2017 Jun 12. Cancer Discov. 2017. PMID: 28607097 No abstract available.
Abstract
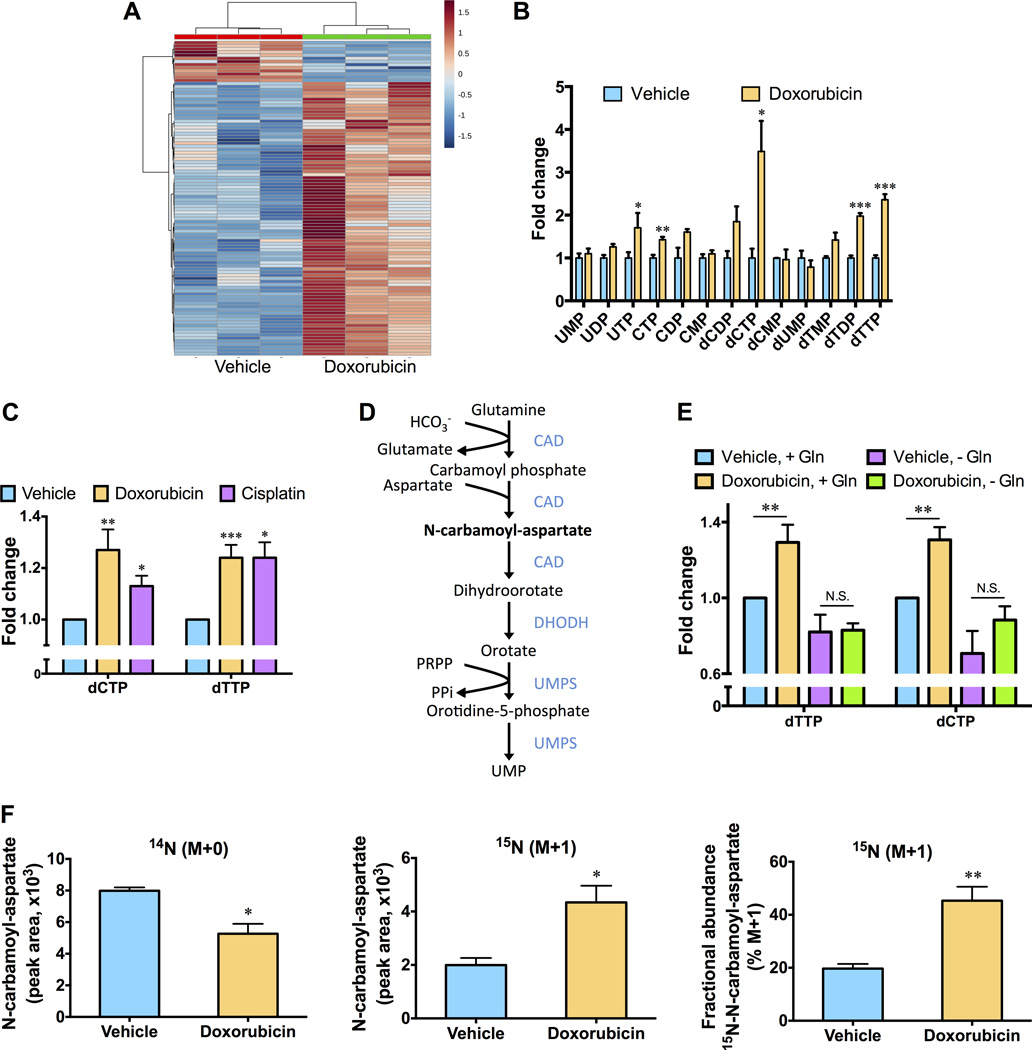
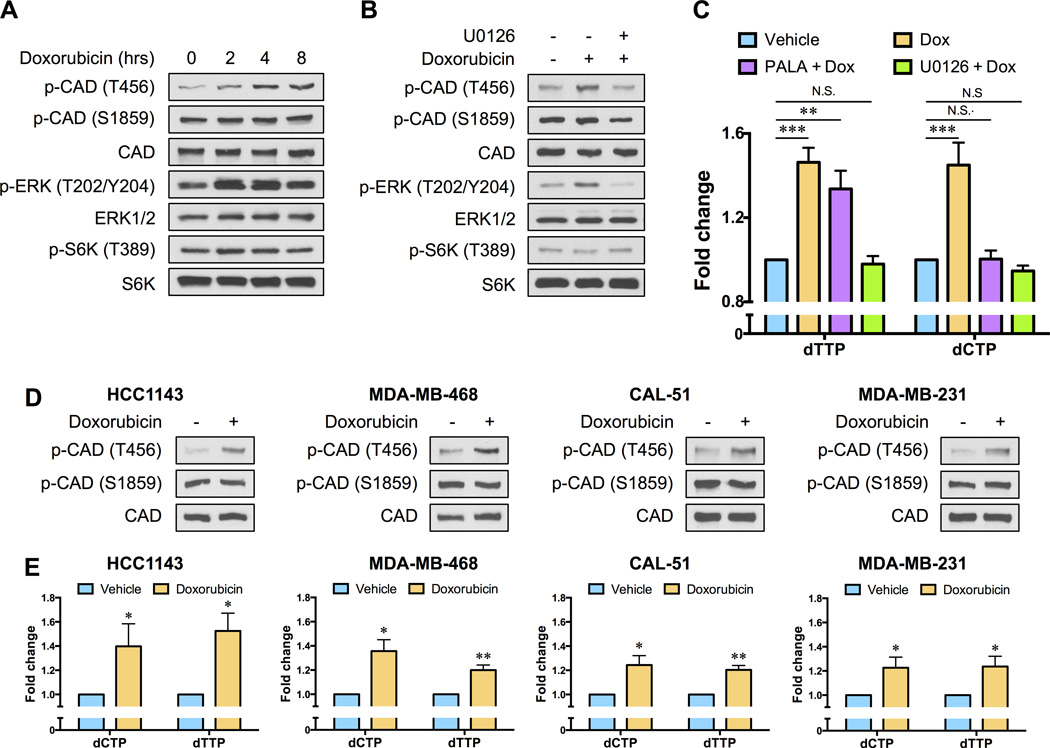
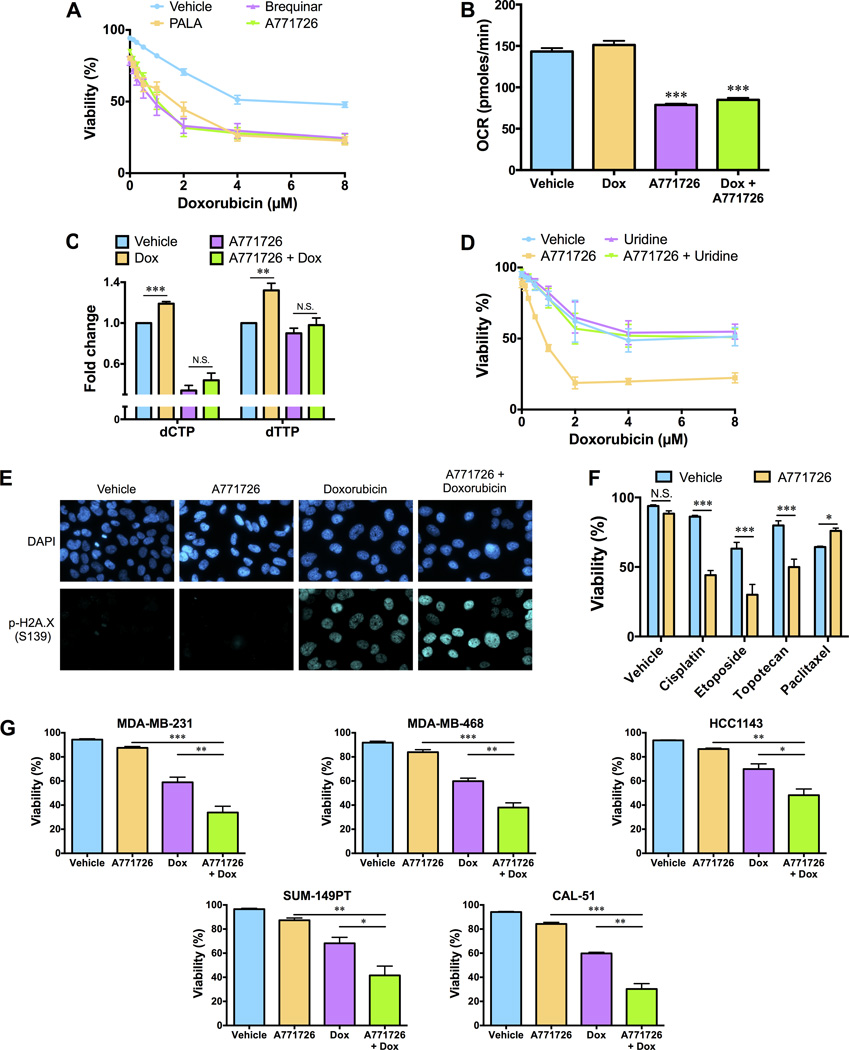
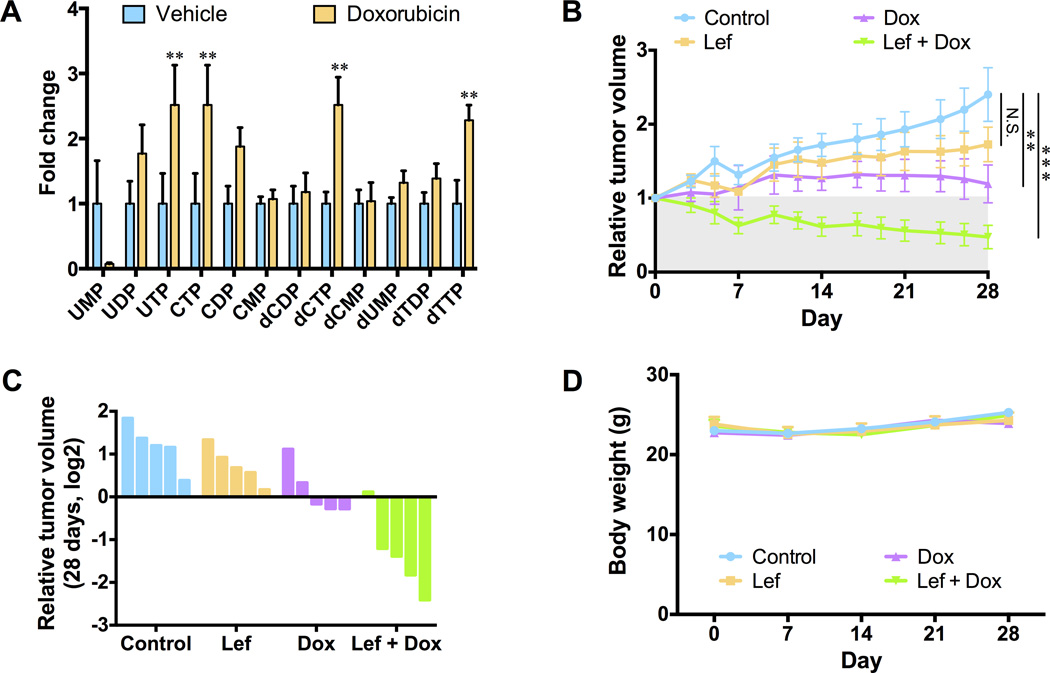
Chemotherapy resistance is a major barrier to the treatment of triple-negative breast cancer (TNBC), and strategies to circumvent resistance are required. Using in vitro and in vivo metabolic profiling of TNBC cells, we show that an increase in the abundance of pyrimidine nucleotides occurs in response to chemotherapy exposure. Mechanistically, elevation of pyrimidine nucleotides induced by chemotherapy is dependent on increased activity of the de novo pyrimidine synthesis pathway. Pharmacologic inhibition of de novo pyrimidine synthesis sensitizes TNBC cells to genotoxic chemotherapy agents by exacerbating DNA damage. Moreover, combined treatment with doxorubicin and leflunomide, a clinically approved inhibitor of the de novo pyrimidine synthesis pathway, induces regression of TNBC xenografts. Thus, the increase in pyrimidine nucleotide levels observed following chemotherapy exposure represents a metabolic vulnerability that can be exploited to enhance the efficacy of chemotherapy for the treatment of TNBC.Significance: The prognosis for patients with TNBC with residual disease after chemotherapy is poor. We find that chemotherapy agents induce adaptive reprogramming of de novo pyrimidine synthesis and show that this response can be exploited pharmacologically, using clinically approved inhibitors of de novo pyrimidine synthesis, to sensitize TNBC cells to chemotherapy. Cancer Discov; 7(4); 391-9. ©2017 AACR.See related article by Mathur et al., p. 380This article is highlighted in the In This Issue feature, p. 339.
©2017 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Guarneri V, Broglio K, Kau SW, Cristofanilli M, Buzdar AU, Valero V, et al. Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J Clin Oncol. 2006;24:1037–1044. - PubMed
-
- Kuerer HM, Newman LA, Smith TL, Ames FC, Hunt KK, Dhingra K, et al. Clinical course of breast cancer patients with complete pathologic primary tumor and axillary lymph node response to doxorubicin-based neoadjuvant chemotherapy. J Clin Oncol. 1999;17:460–469. - PubMed
-
- Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275–1281. - PubMed
-
- Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384:164–172. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
