

Mutations in SLC25A22: hyperprolinaemia, vacuolated fibroblasts and presentation with developmental delay
- PMID: 28255779
- PMCID: PMC5393281
- DOI: 10.1007/s10545-017-0025-7
Mutations in SLC25A22: hyperprolinaemia, vacuolated fibroblasts and presentation with developmental delay
Abstract
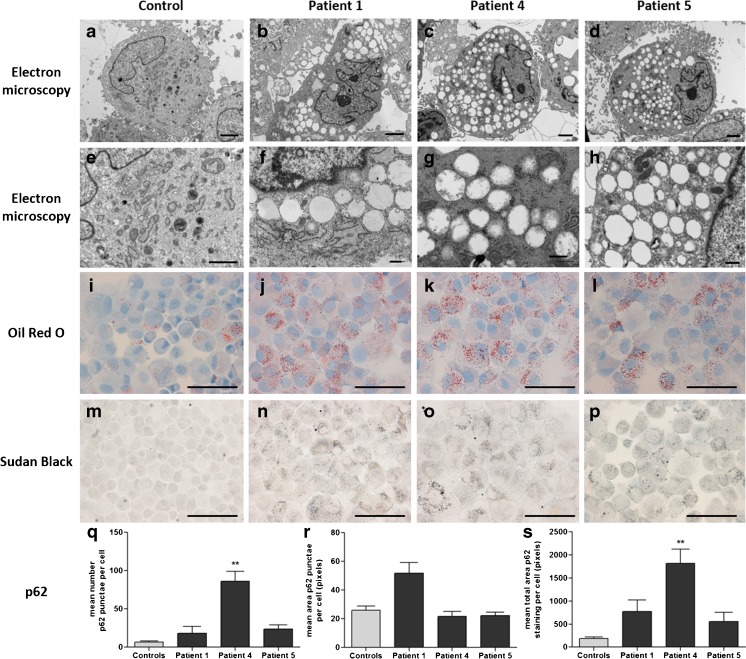
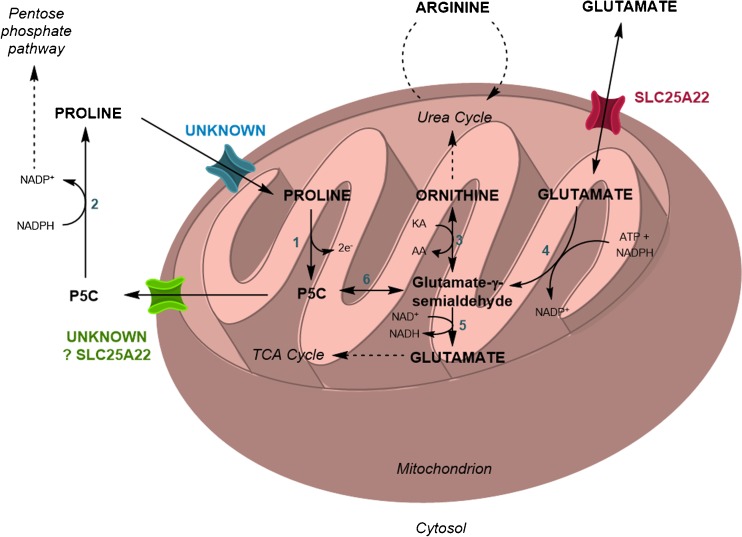
Mutations in SLC25A22 are known to cause neonatal epileptic encephalopathy and migrating partial seizures in infancy. Using whole exome sequencing we identified four novel SLC25A22 mutations in six children from three families. Five patients presented clinical features similar to those in the literature including hypotonia, refractory neonatal-onset seizures and developmental delay. However, the sixth patients presented atypically with isolated developmental delay, developing late-onset (absence) seizures only at 7 years of age. Abnormal metabolite levels have not been documented in the nine patients described previously. One patient in our series was referred to the metabolic clinic because of persistent hyperprolinaemia and another three had raised plasma proline when tested. Analysis of the post-prandial plasma amino acid response in one patient showed abnormally high concentrations of several amino acids. This suggested that, in the fed state, when amino acids are the preferred fuel for the liver, trans-deamination of amino acids requires transportation of glutamate into liver mitochondria by SLC25A22 for deamination by glutamate dehydrogenase; SLC25A22 is an important mitochondrial glutamate transporter in liver as well as in brain. Electron microscopy of patient fibroblasts demonstrated widespread vacuolation containing neutral and phospho-lipids as demonstrated by Oil Red O and Sudan Black tinctorial staining; this might be explained by impaired activity of the proline/pyrroline-5-carboxylate (P5C) shuttle if SLC25A22 transports pyrroline-5-carboxylate/glutamate-γ-semialdehyde as well as glutamate.
Conflict of interest statement
Funding
PBM, SR and PTC are supported by Great Ormond Street Hospital Children’s Charity (GOSHCC). PG is a Welcome Trust Senior Research fellow. This project was funded by grants from the University College London Impact Award and GOSHCC Metabolic Fund. GOSgene is supported by the NIHR BRC at GOSH for Children NHS Foundation Trust and UCL Institute of Child Health. Views expressed are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
Conflict of interest
None.
Use of laboratory animals
Not applicable
Figures

References
-
- Baumgartner MR, Rabier D, Nassogne MC, Dufier JL, Padovani JP, Kamoun P, et al. Delta1-pyrroline-5-carboxylate synthase deficiency: neurodegeneration, cataracts and connective tissue manifestations combined with hyperammonaemia and reduced ornithine, citrulline, arginine and proline. Eur J Pediatr. 2005;164:31–36. doi: 10.1007/s00431-004-1545-3. - DOI - PubMed
-
- Cohen R, Basel-Vanagaite L, Goldberg-Stern H, Halevy A, Shuper A, Feingold-Zadok M, et al. Two siblings with early infantile myoclonic encephalopathy due to mutation in the gene encoding mitochondrial glutamate/H+ symporter SLC25A22. Eur J Paediatr Neurol. 2014;18(6):801–805. doi: 10.1016/j.ejpn.2014.06.007. - DOI - PubMed
-
- Fiermonte G, Palmieri L, Todisco S, Agrimi G, Palmieri F, Walker JE. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2002;277(22):19289–19294. doi: 10.1074/jbc.M201572200. - DOI - PubMed
URLs
-
- Human Gene Mutation Database* (http://www.biobase-international.com)
-
- SIFT* (http://sift.bii.a-star.edu.sg/)
-
- PolyPhen-2* (http://genetics.bwh.harvard.edu/pph2/)
-
- ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
