

Tissue-specific tumorigenesis: context matters
- PMID: 28256574
- PMCID: PMC5823237
- DOI: 10.1038/nrc.2017.5
Tissue-specific tumorigenesis: context matters
Abstract
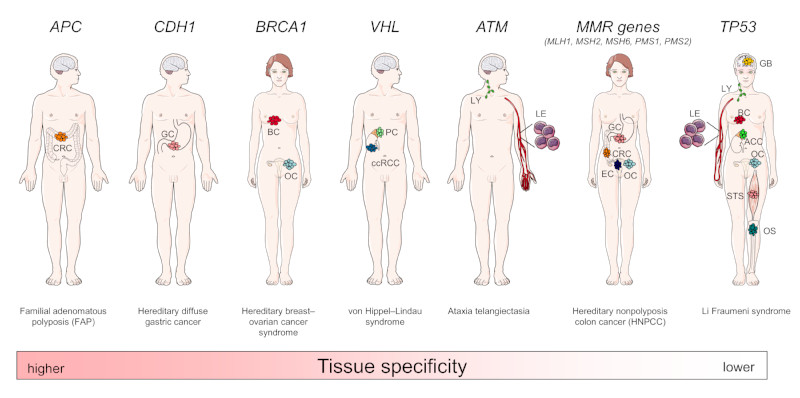
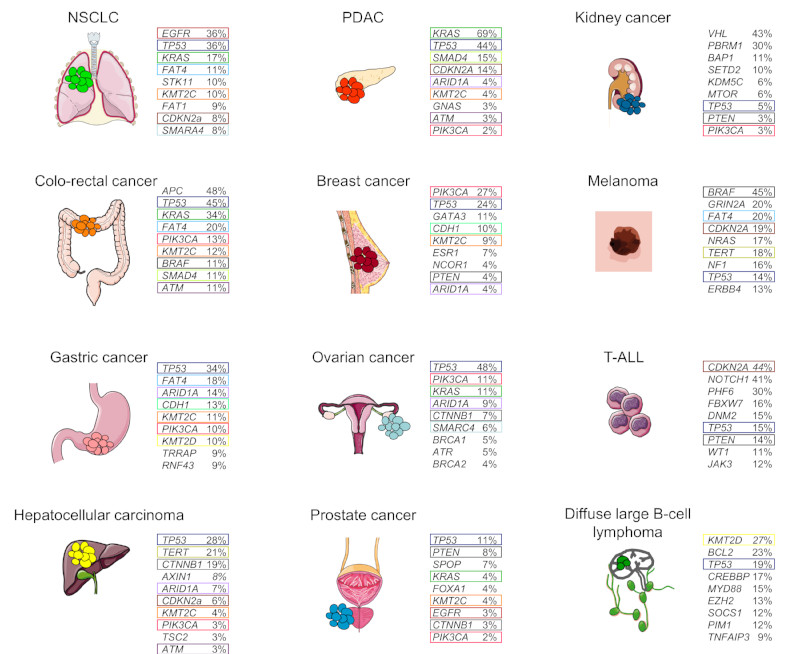
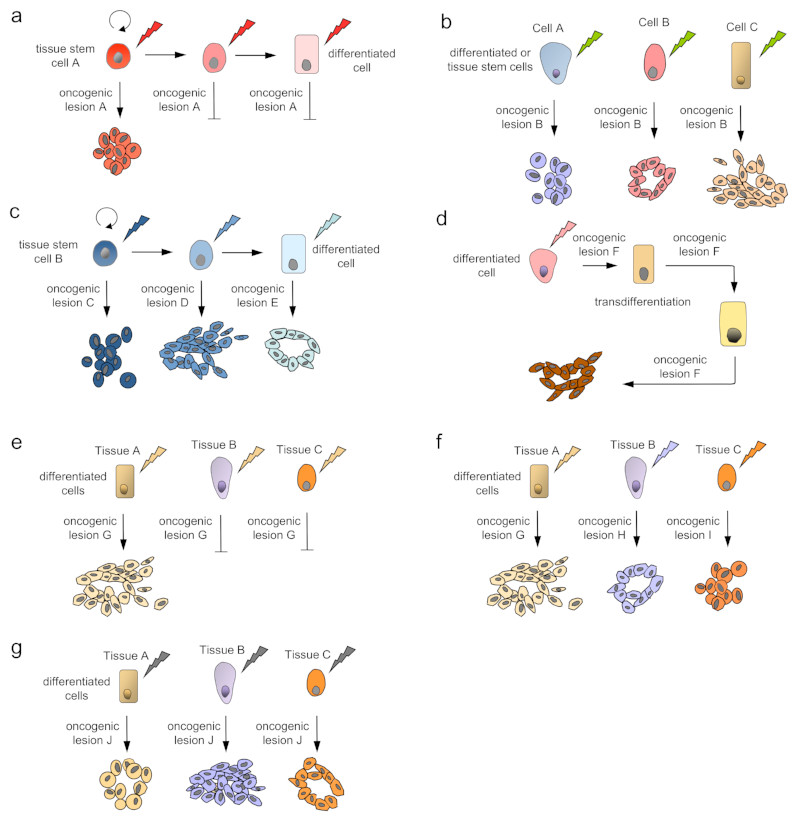
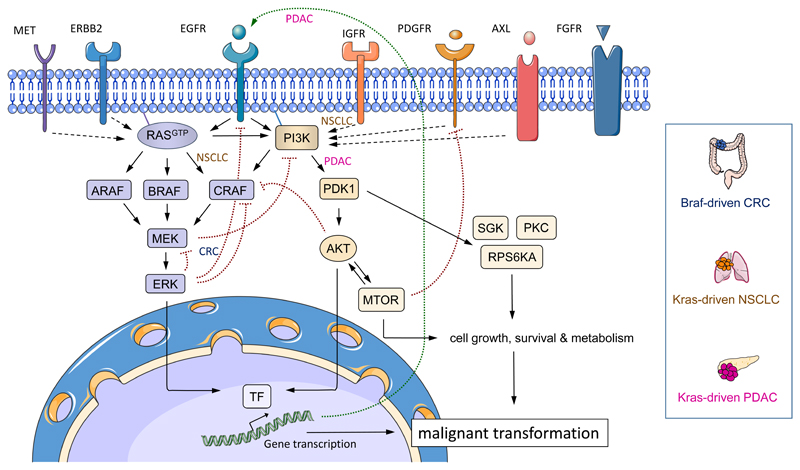
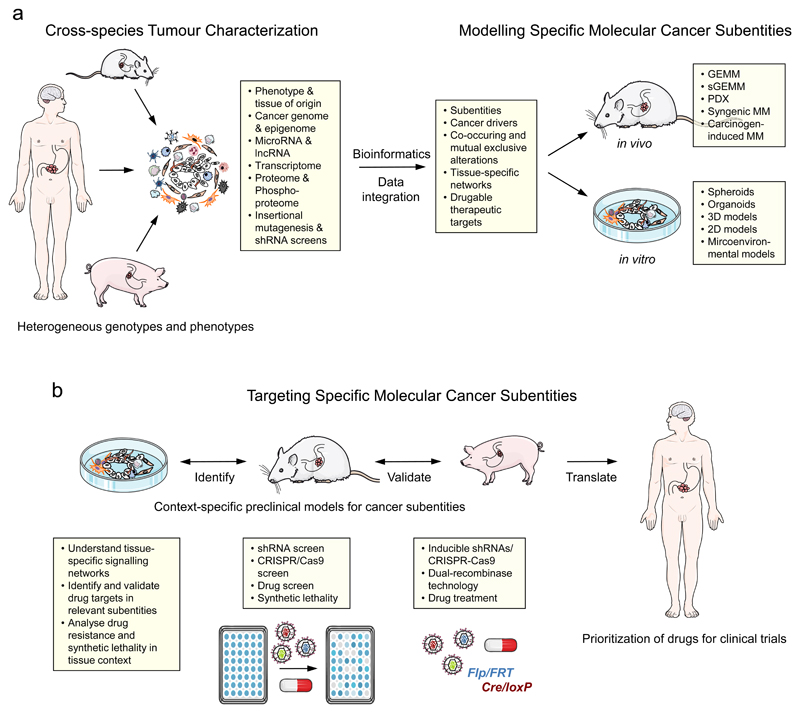
How can we treat cancer more effectively? Traditionally, tumours from the same anatomical site are treated as one tumour entity. This concept has been challenged by recent breakthroughs in cancer genomics and translational research that have enabled molecular tumour profiling. The identification and validation of cancer drivers that are shared between different tumour types, spurred the new paradigm to target driver pathways across anatomical sites by off-label drug use, or within so-called basket or umbrella trials which are designed to test whether molecular alterations in one tumour entity can be extrapolated to all others. However, recent clinical and preclinical studies suggest that there are tissue- and cell type-specific differences in tumorigenesis and the organization of oncogenic signalling pathways. In this Opinion article, we focus on the molecular, cellular, systemic and environmental determinants of organ-specific tumorigenesis and the mechanisms of context-specific oncogenic signalling outputs. Investigation, recognition and in-depth biological understanding of these differences will be vital for the design of next-generation clinical trials and the implementation of molecularly guided cancer therapies in the future.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Dancey JE, Bedard PL, Onetto N, Hudson TJ. The genetic basis for cancer treatment decisions. Cell. 2012;148:409–420. - PubMed
-
- Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. - PubMed
-
- Demetri GD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. - PubMed
-
- Rosell R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
