

Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1
- PMID: 28257700
- PMCID: PMC5507214
- DOI: 10.1016/j.molcel.2017.01.029
Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1
Abstract
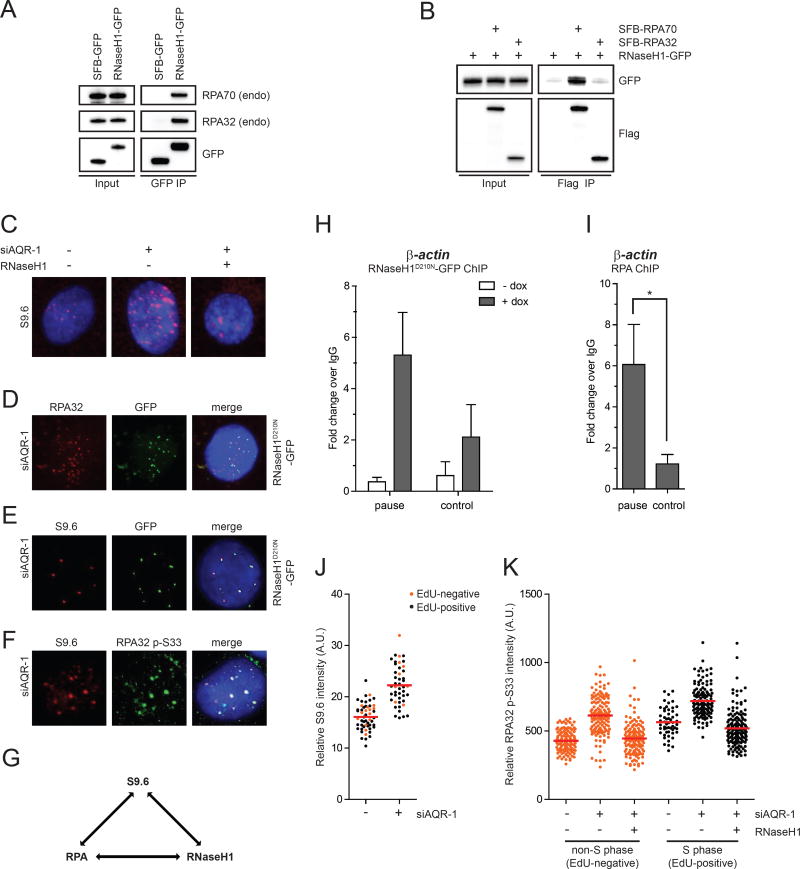
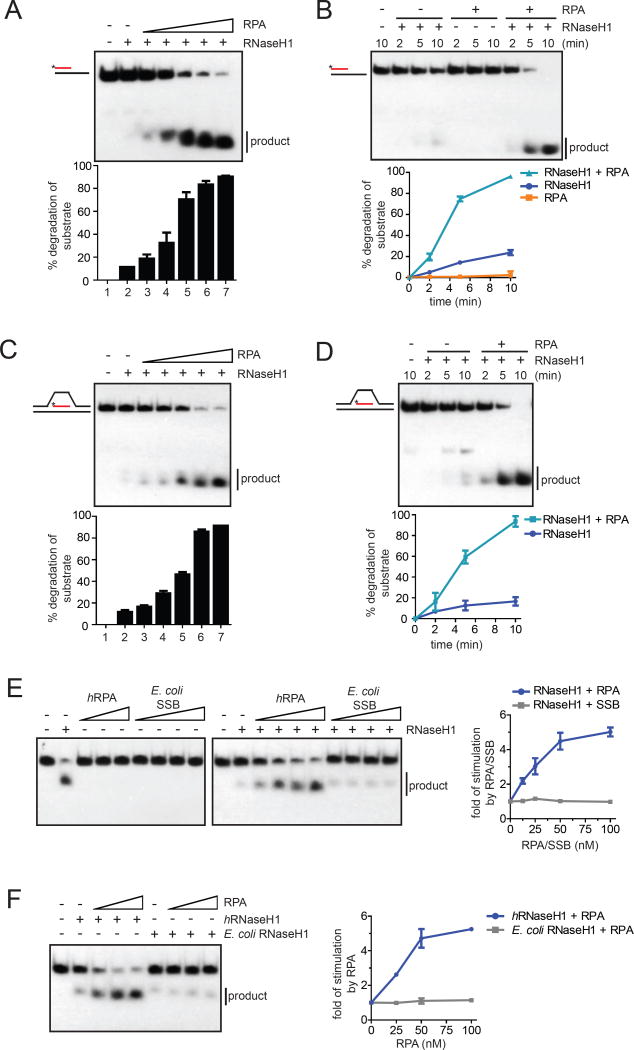
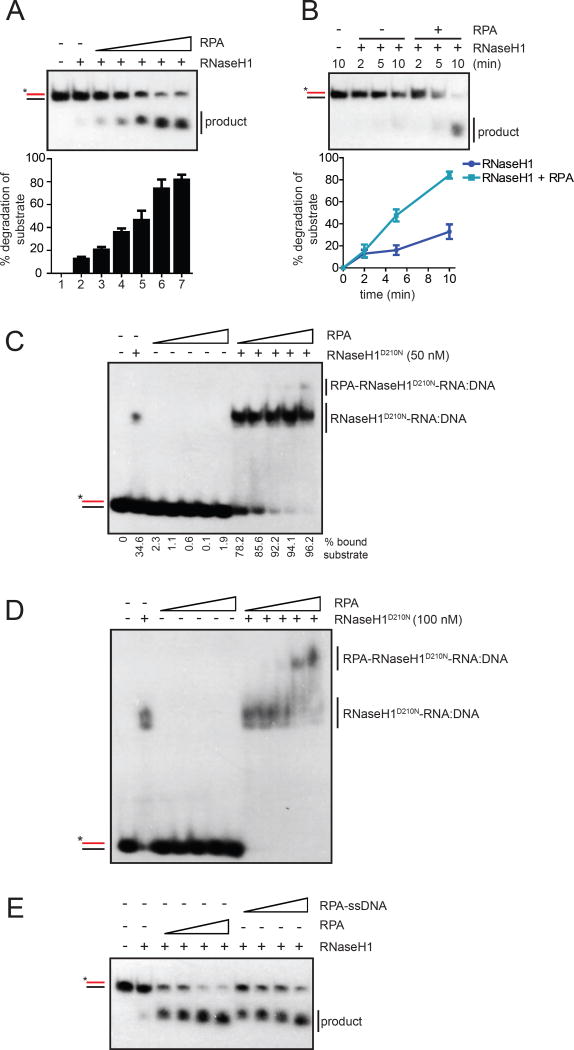
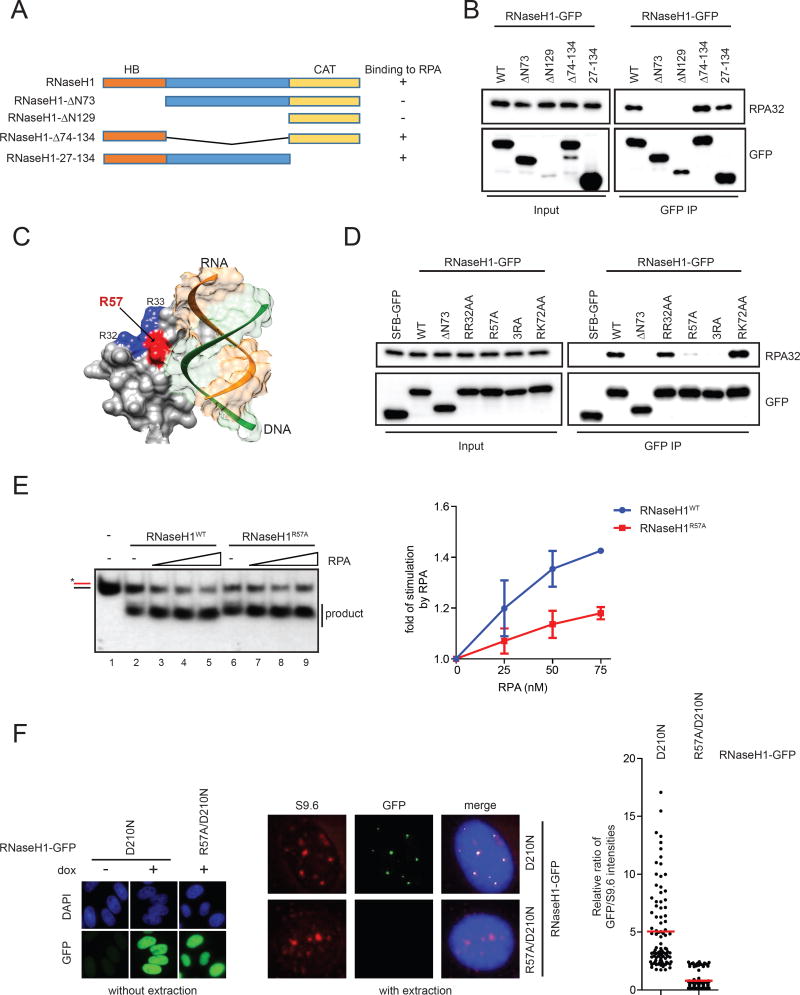
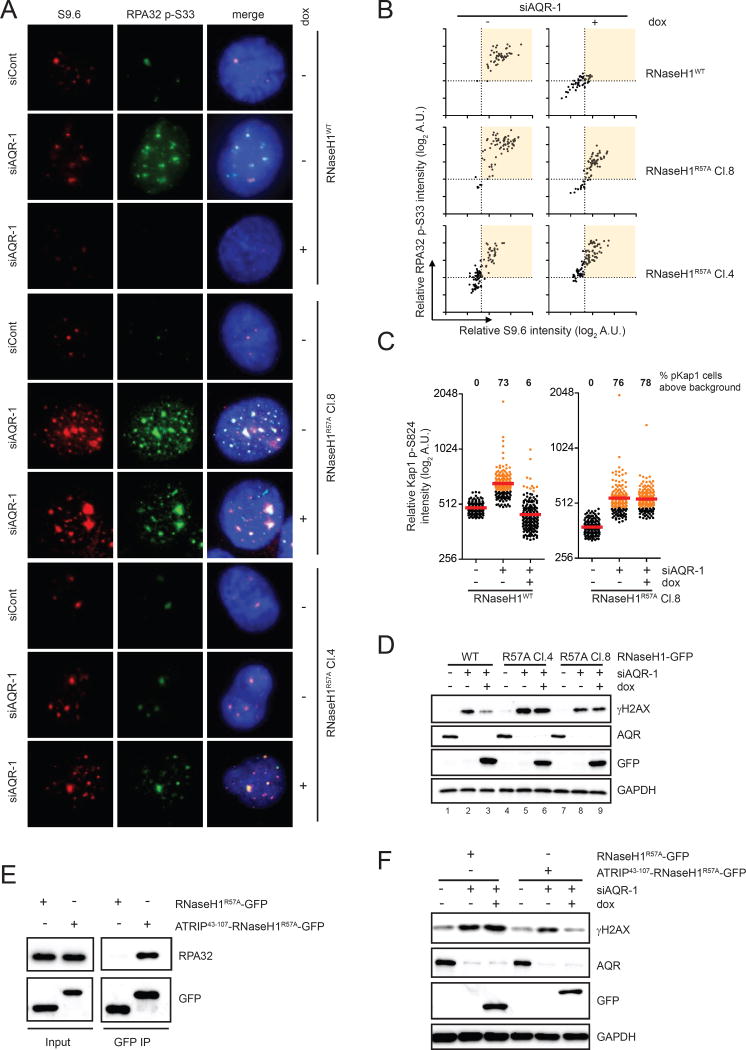
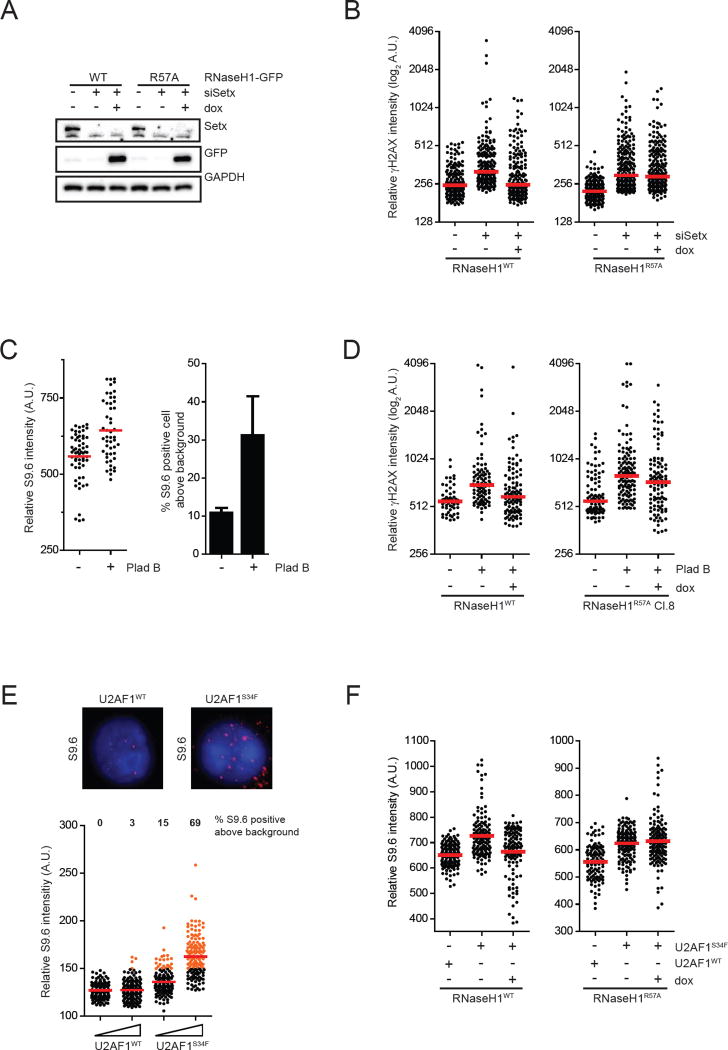
R loop, a transcription intermediate containing RNA:DNA hybrids and displaced single-stranded DNA (ssDNA), has emerged as a major source of genomic instability. RNaseH1, which cleaves the RNA in RNA:DNA hybrids, plays an important role in R loop suppression. Here we show that replication protein A (RPA), an ssDNA-binding protein, interacts with RNaseH1 and colocalizes with both RNaseH1 and R loops in cells. In vitro, purified RPA directly enhances the association of RNaseH1 with RNA:DNA hybrids and stimulates the activity of RNaseH1 on R loops. An RPA binding-defective RNaseH1 mutant is not efficiently stimulated by RPA in vitro, fails to accumulate at R loops in cells, and loses the ability to suppress R loops and associated genomic instability. Thus, in addition to sensing DNA damage and replication stress, RPA is a sensor of R loops and a regulator of RNaseH1, extending the versatile role of RPA in suppression of genomic instability.
Keywords: R loop; RNase H1; RPA; genome instability; splicing inhibitor; splicing mutation; ssDNA.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511:362–365. - PubMed
-
- Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL, Carrico RJ. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J. Immunol. Methods. 1986;89:123–130. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
