

Effect of α7 nicotinic acetylcholine receptor activation on cardiac fibroblasts: a mechanism underlying RV fibrosis associated with cigarette smoke exposure
- PMID: 28258105
- PMCID: PMC5451597
- DOI: 10.1152/ajplung.00393.2016
Effect of α7 nicotinic acetylcholine receptor activation on cardiac fibroblasts: a mechanism underlying RV fibrosis associated with cigarette smoke exposure
Abstract
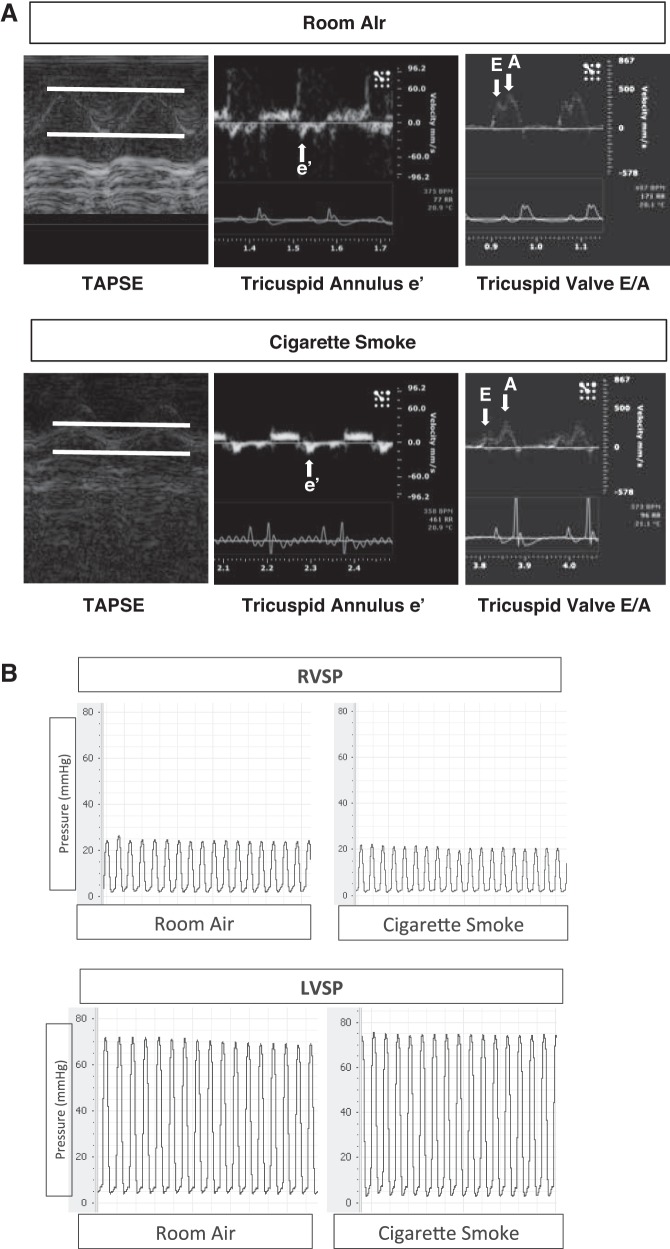
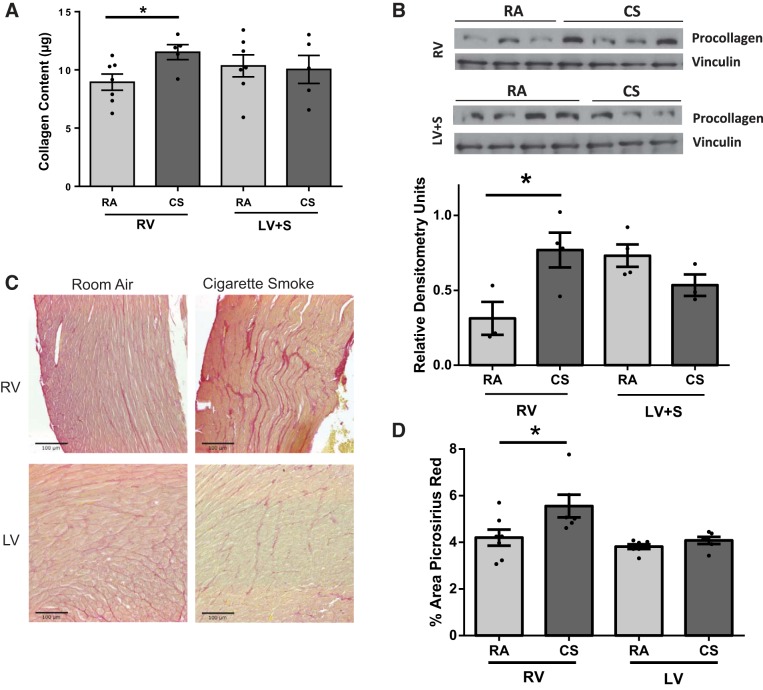
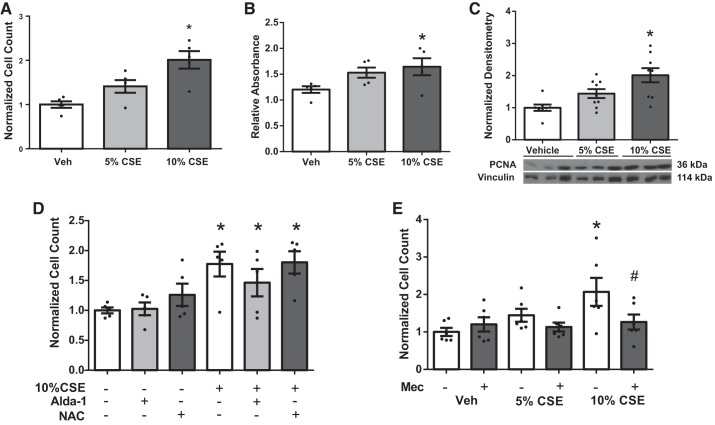
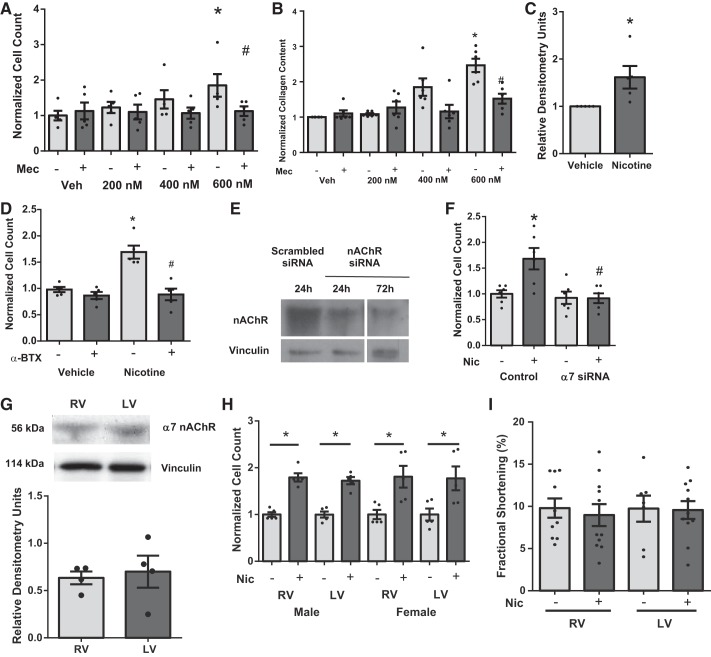
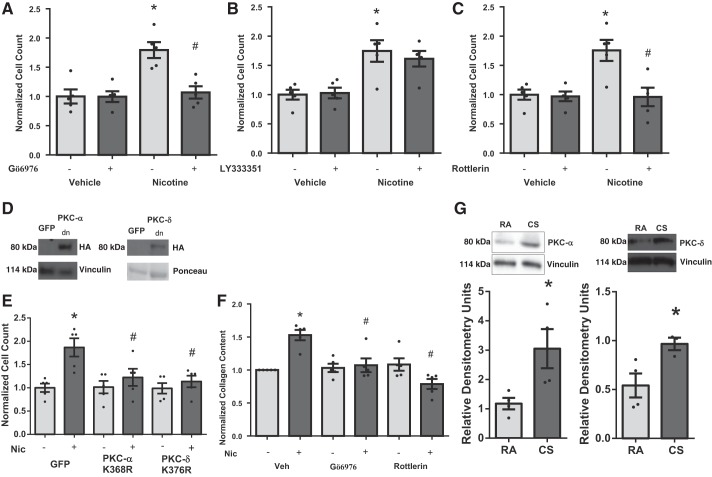
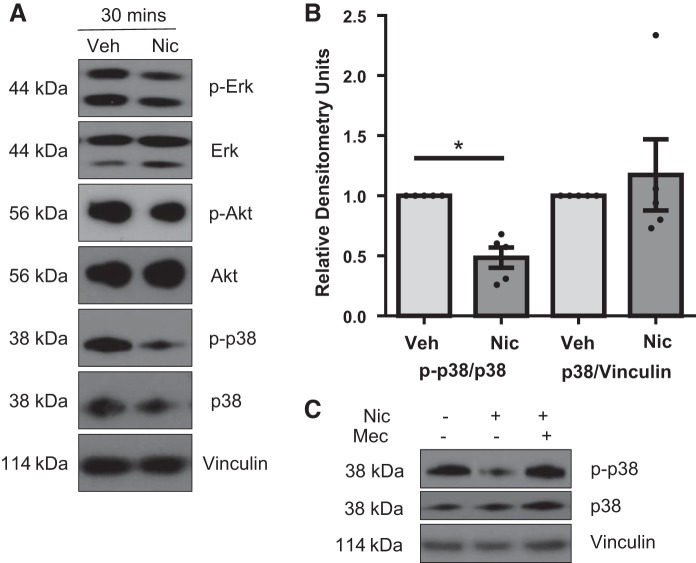
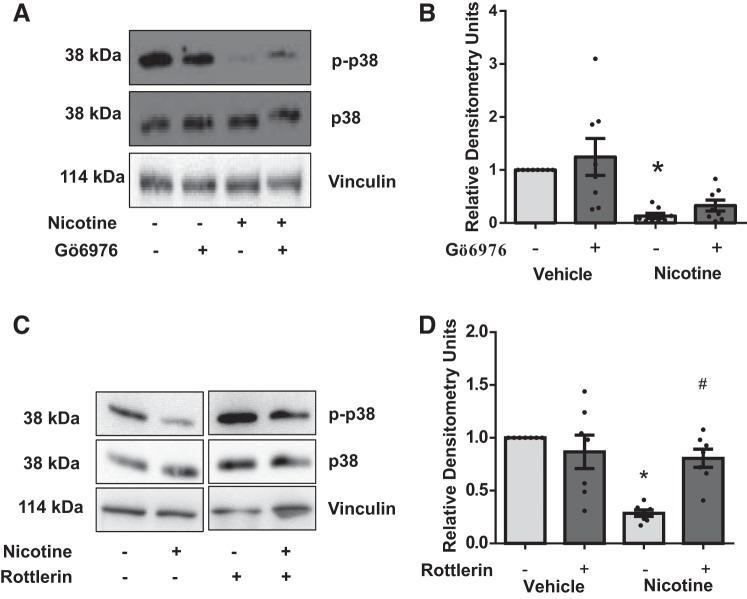
Right ventricular (RV) dysfunction is associated with numerous smoking-related illnesses, including chronic obstructive pulmonary disease (COPD), in which it is present even in the absence of pulmonary hypertension. It is unknown whether exposure to cigarette smoke (CS) has direct effects on RV function and cardiac fibroblast (CF) proliferation or collagen synthesis. In this study, we evaluated cardiac function and fibrosis in mice exposed to CS and determined mechanisms of smoke-induced changes in CF signaling and fibrosis. AKR mice were exposed to CS for 6 wk followed by echocardiography and evaluation of cardiac hypertrophy, collagen content, and pulmonary muscularization. Proliferation and collagen content were evaluated in primary isolated rat CFs exposed to CS extract (CSE) or nicotine. Markers of cell proliferation, fibrosis, and proliferative signaling were determined by immunoblot or Sircol collagen assay. Mice exposed to CS had significantly decreased RV function, as determined by tricuspid annular plane systolic excursion. There were no changes in left ventricular parameters. RV collagen content was significantly elevated, but there was no change in RV hypertrophy or pulmonary vascular muscularization. CSE directly increased CF proliferation and collagen content in CF. Nicotine alone reproduced these effects. CSE and nicotine-induced fibroblast proliferation and collagen content were mediated through α7 nicotinic acetylcholine receptors and were dependent on PKC-α, PKC-δ, and reduced p38-MAPK phosphorylation. CS and nicotine have direct effects on CFs to induce proliferation and fibrosis, which may negatively affect right heart function.
Keywords: chronic obstructive pulmonary disease; fibrosis; nicotine; nicotinic acetylcholine receptor; right ventricle.
Figures
References
-
- Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest 111: 1475–1486, 2003. doi:10.1172/JCI200317295. - DOI - PMC - PubMed
-
- Chichger H, Vang A, O’Connell KA, Zhang P, Mende U, Harrington EO, Choudhary G. PKC d and bII regulate angiotensin II-mediated fibrosis through p38: a mechanism of RV fibrosis in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L827–L836, 2015. doi:10.1152/ajplung.00184.2014. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials