

A Pooled Sequencing Approach Identifies a Candidate Meiotic Driver in Drosophila
- PMID: 28258181
- PMCID: PMC5419488
- DOI: 10.1534/genetics.116.197335
A Pooled Sequencing Approach Identifies a Candidate Meiotic Driver in Drosophila
Abstract
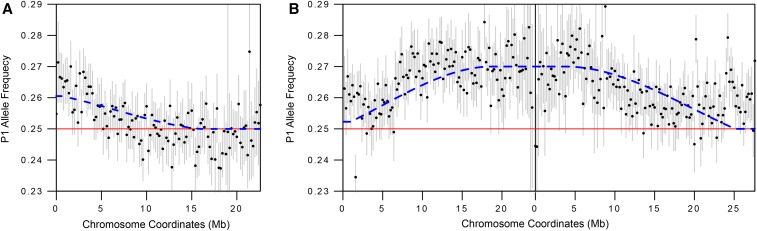
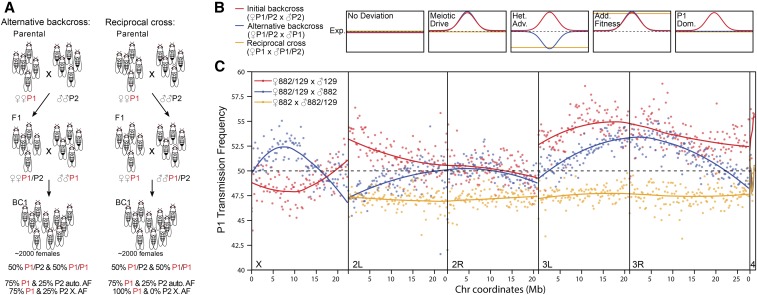
Meiotic drive occurs when a selfish element increases its transmission frequency above the Mendelian ratio by hijacking the asymmetric divisions of female meiosis. Meiotic drive causes genomic conflict and potentially has a major impact on genome evolution, but only a few drive loci of large effect have been described. New methods to reliably detect meiotic drive are therefore needed, particularly for discovering moderate-strength drivers that are likely to be more prevalent in natural populations than strong drivers. Here, we report an efficient method that uses sequencing of large pools of backcross (BC1) progeny to test for deviations from Mendelian segregation genome-wide with single-nucleotide polymorphisms (SNPs) that distinguish the parental strains. We show that meiotic drive can be detected by a characteristic pattern of decay in distortion of SNP frequencies, caused by recombination unlinking the driver from distal loci. We further show that control crosses allow allele-frequency distortion caused by meiotic drive to be distinguished from distortion resulting from developmental effects. We used this approach to test whether chromosomes with extreme telomere-length differences segregate at Mendelian ratios, as telomeric regions are a potential hotspot for meiotic drive due to their roles in meiotic segregation and multiple observations of high rates of telomere sequence evolution. Using four different pairings of long and short telomere strains, we find no evidence that extreme telomere-length variation causes meiotic drive in Drosophila However, we identify one candidate meiotic driver in a centromere-linked region that shows an ∼8% increase in transmission frequency, corresponding to a ∼54:46 segregation ratio. Our results show that candidate meiotic drivers of moderate strength can be readily detected and localized in pools of BC1 progeny.
Keywords: Drosophila; centromeres; drive; meiotic; telomeres.
Copyright © 2017 by the Genetics Society of America.
Figures
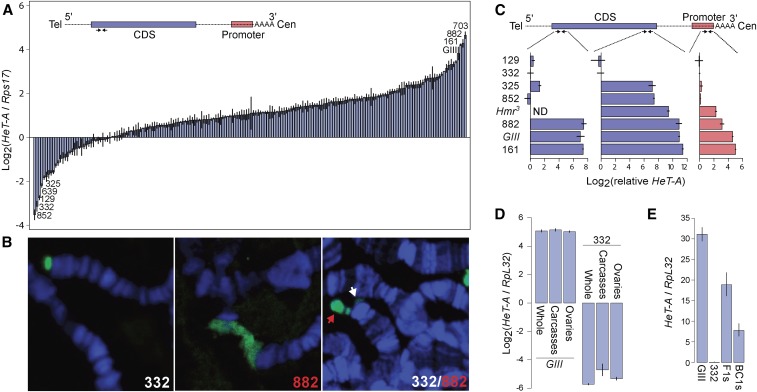
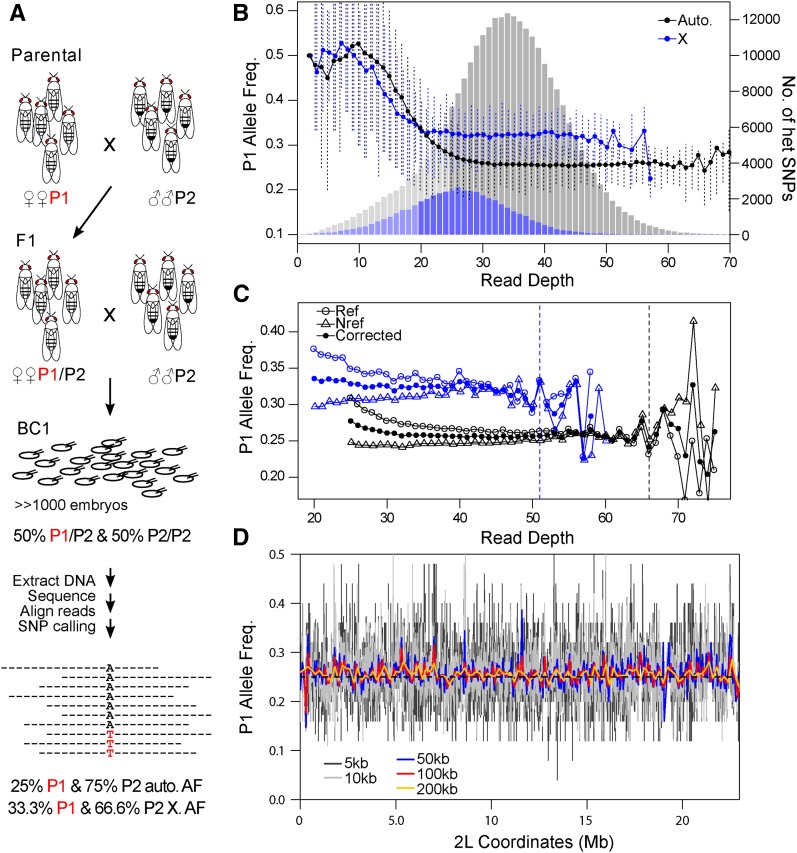
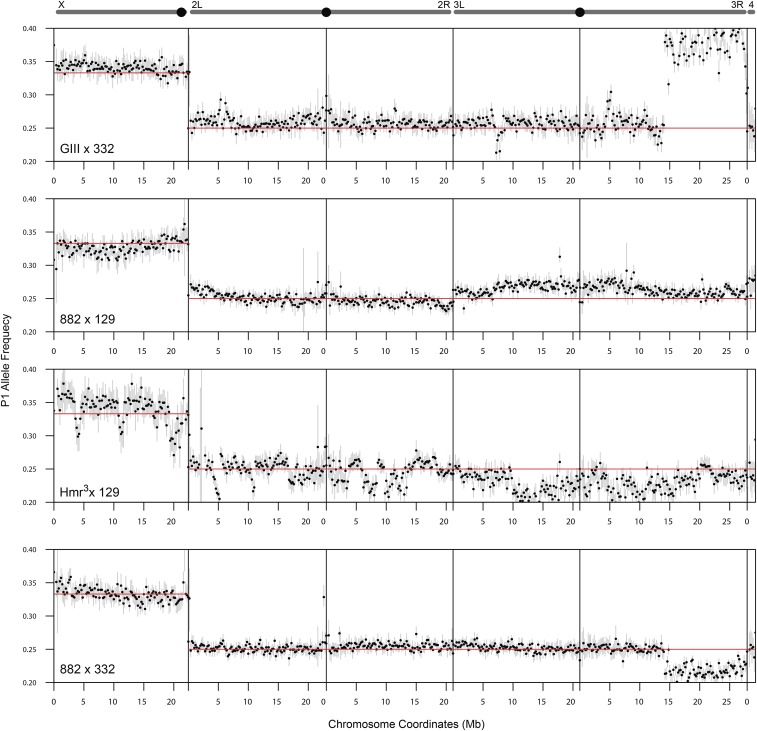
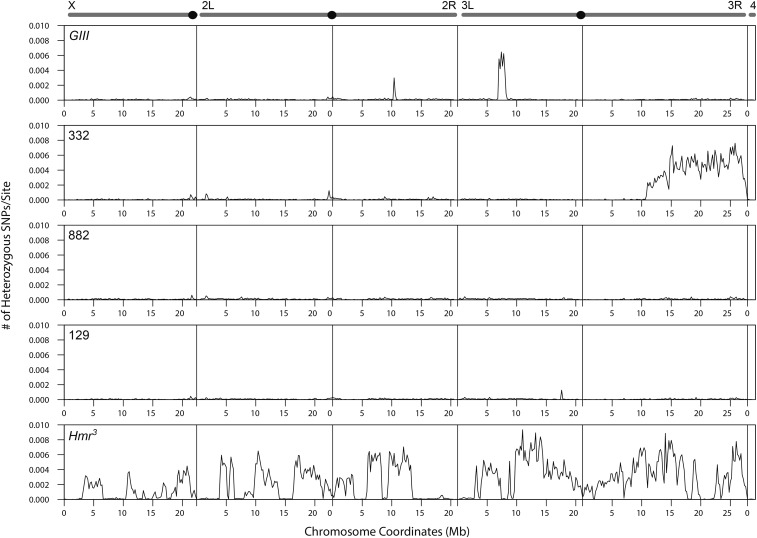
References
-
- Bélanger S., Clermont I., Esteves P., Belzile F., 2016. Extent and overlap of segregation distortion regions in 12 barley crosses determined via a Pool-GBS approach. Theor. Appl. Genet. 129: 1393–1404. - PubMed
-
- Blumenstiel J. P., 2011. Evolutionary dynamics of transposable elements in a small RNA world. Trends Genet. 27: 23–31. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
