

Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome
- PMID: 28260531
- PMCID: PMC5338099
- DOI: 10.1186/s13059-017-1158-6
Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome
Abstract
Background: Structural variation (SV) influences genome organization and contributes to human disease. However, the complete mutational spectrum of SV has not been routinely captured in disease association studies.
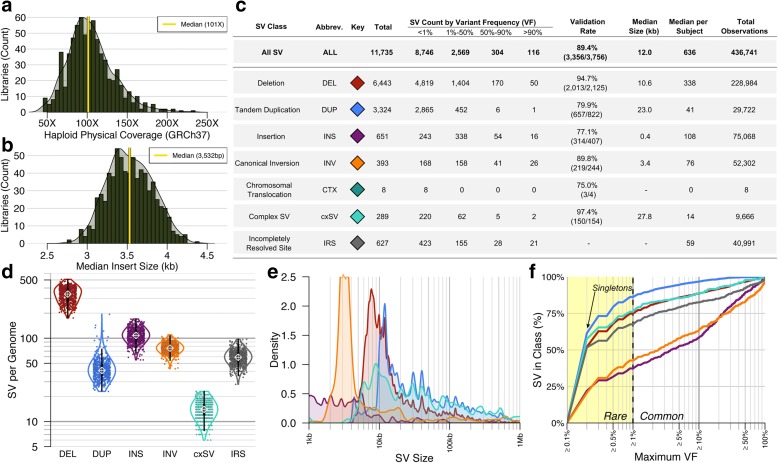
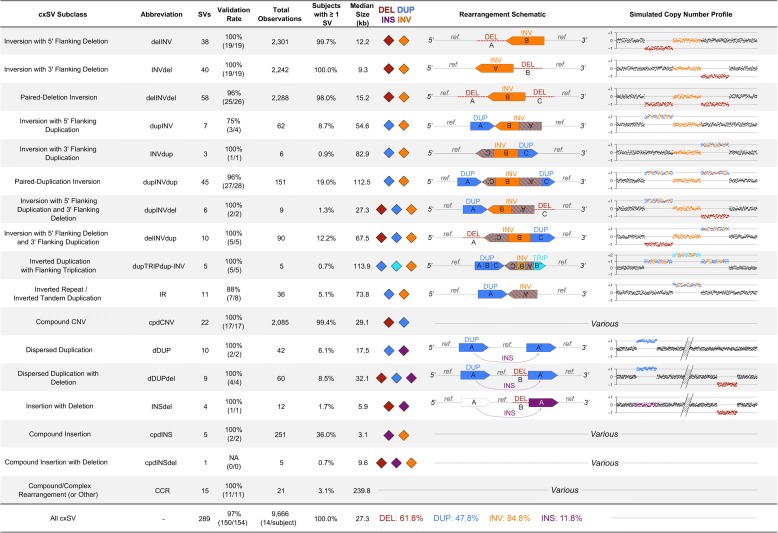
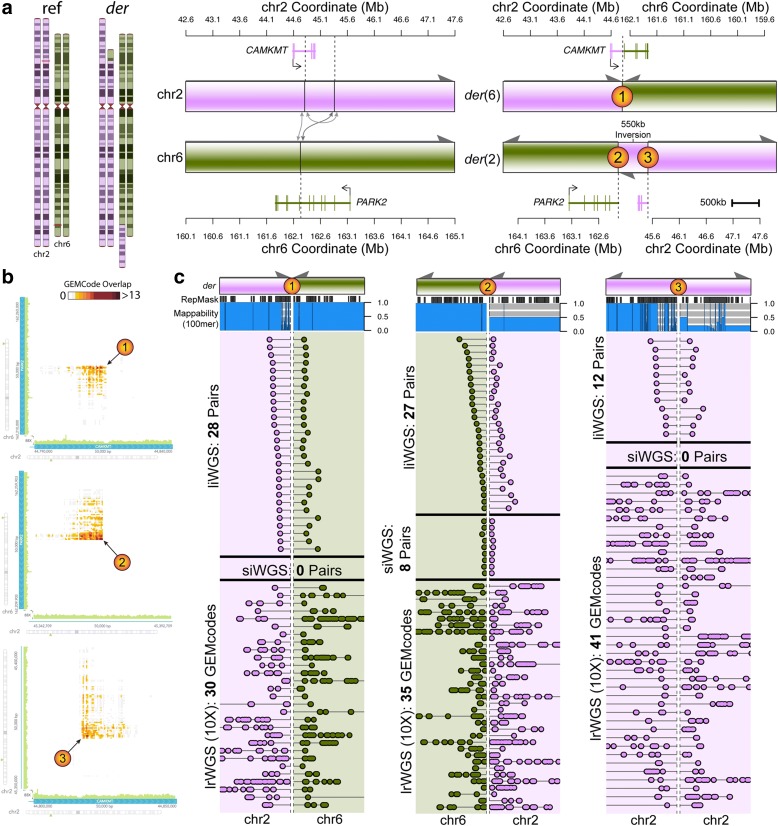
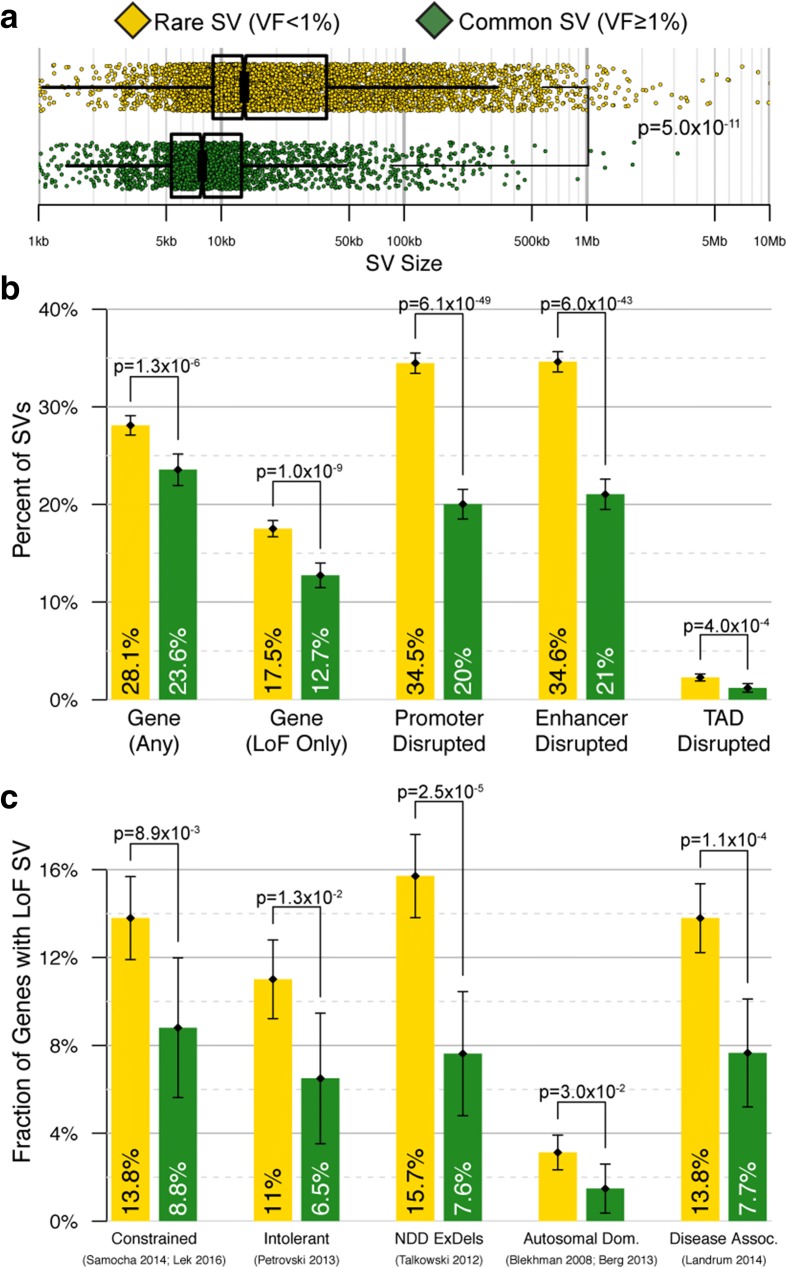
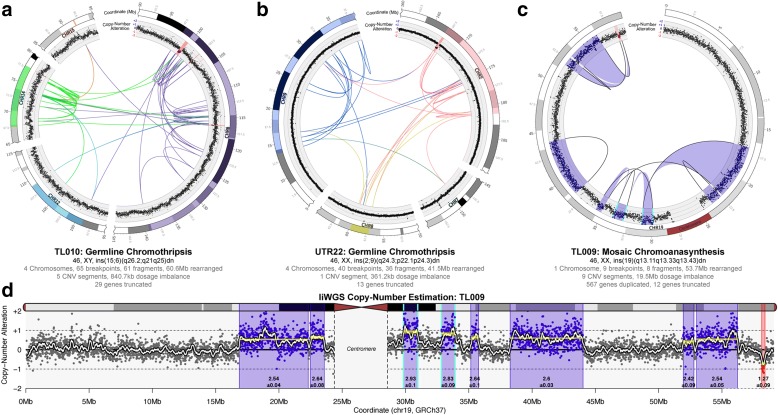
Results: We sequenced 689 participants with autism spectrum disorder (ASD) and other developmental abnormalities to construct a genome-wide map of large SV. Using long-insert jumping libraries at 105X mean physical coverage and linked-read whole-genome sequencing from 10X Genomics, we document seven major SV classes at ~5 kb SV resolution. Our results encompass 11,735 distinct large SV sites, 38.1% of which are novel and 16.8% of which are balanced or complex. We characterize 16 recurrent subclasses of complex SV (cxSV), revealing that: (1) cxSV are larger and rarer than canonical SV; (2) each genome harbors 14 large cxSV on average; (3) 84.4% of large cxSVs involve inversion; and (4) most large cxSV (93.8%) have not been delineated in previous studies. Rare SVs are more likely to disrupt coding and regulatory non-coding loci, particularly when truncating constrained and disease-associated genes. We also identify multiple cases of catastrophic chromosomal rearrangements known as chromoanagenesis, including somatic chromoanasynthesis, and extreme balanced germline chromothripsis events involving up to 65 breakpoints and 60.6 Mb across four chromosomes, further defining rare categories of extreme cxSV.
Conclusions: These data provide a foundational map of large SV in the morbid human genome and demonstrate a previously underappreciated abundance and diversity of cxSV that should be considered in genomic studies of human disease.
Keywords: Autism; Chromoanagenesis; Chromothripsis; Complex chromosomal rearrangement; Copynumber variation; Germline mutation; Inversion; Neurodevelopmental disorders; Structural variation; Whole-genome sequencing.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
