

Variability of Protein Structure Models from Electron Microscopy
- PMID: 28262392
- PMCID: PMC5382112
- DOI: 10.1016/j.str.2017.02.004
Variability of Protein Structure Models from Electron Microscopy
Abstract
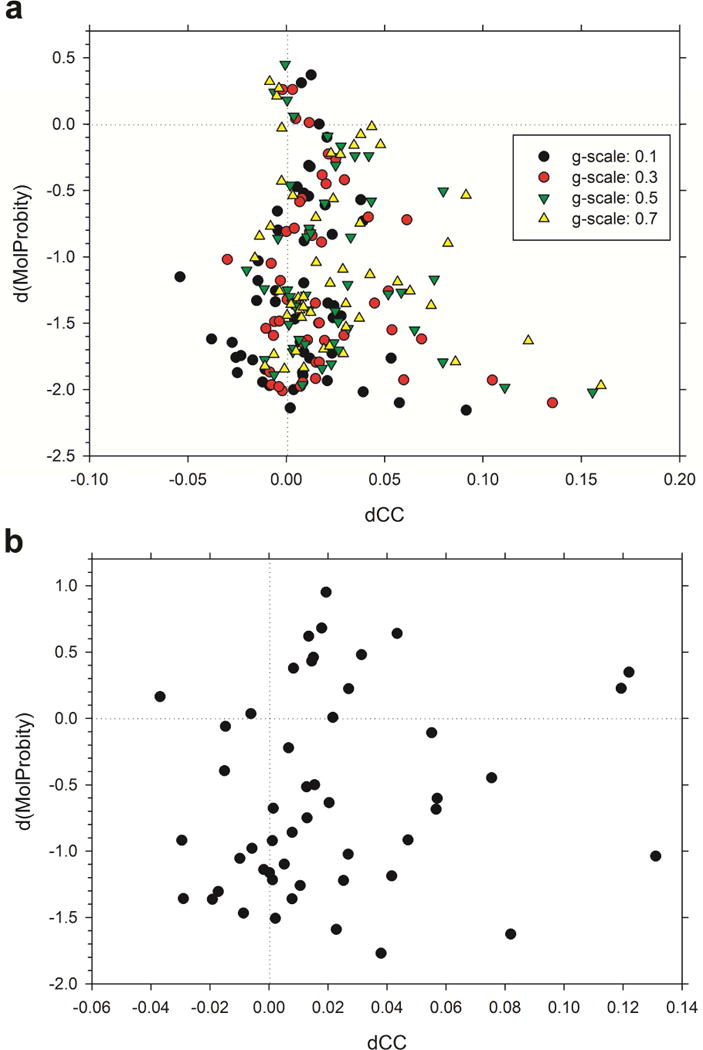
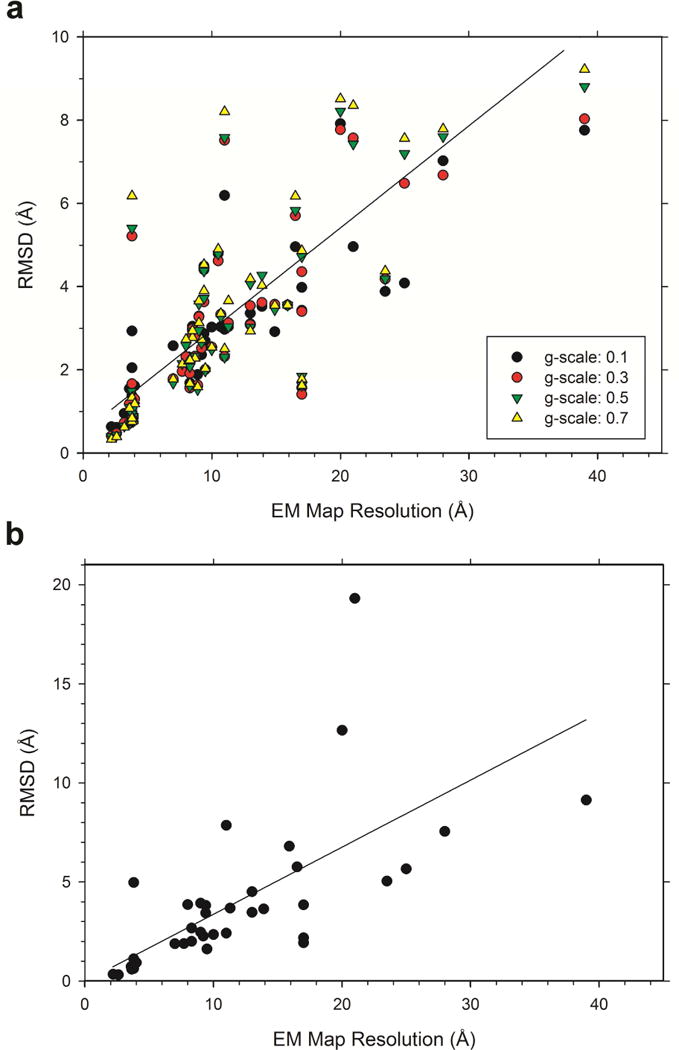
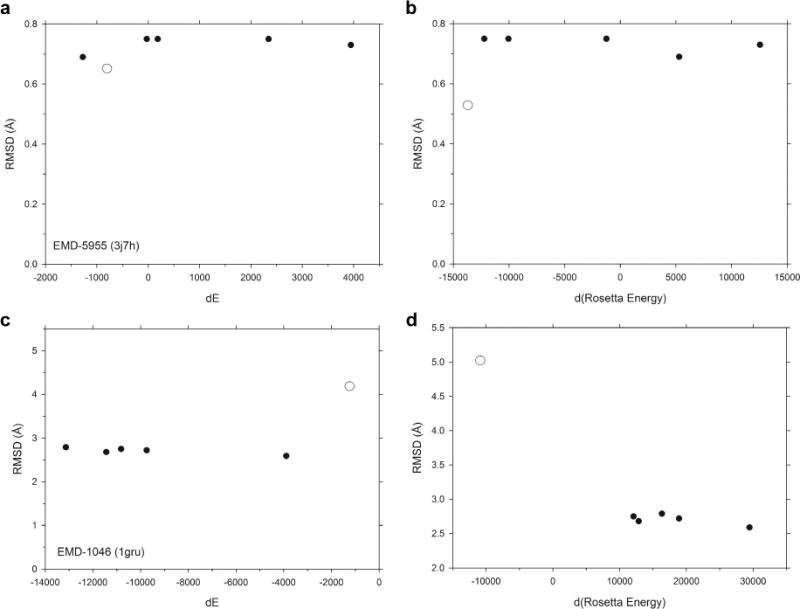
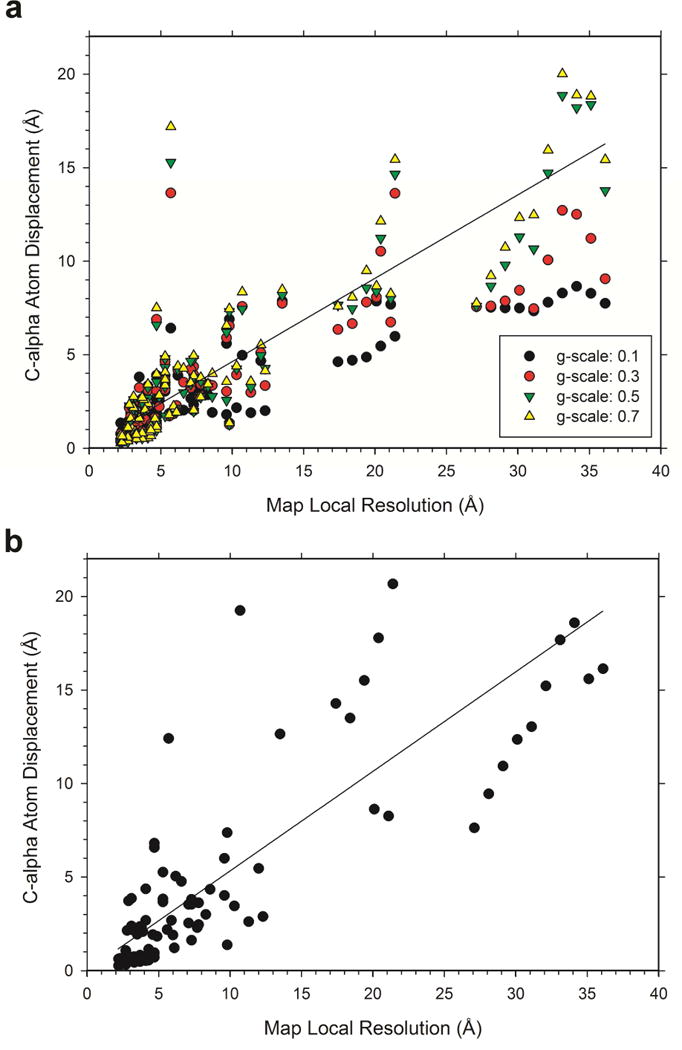
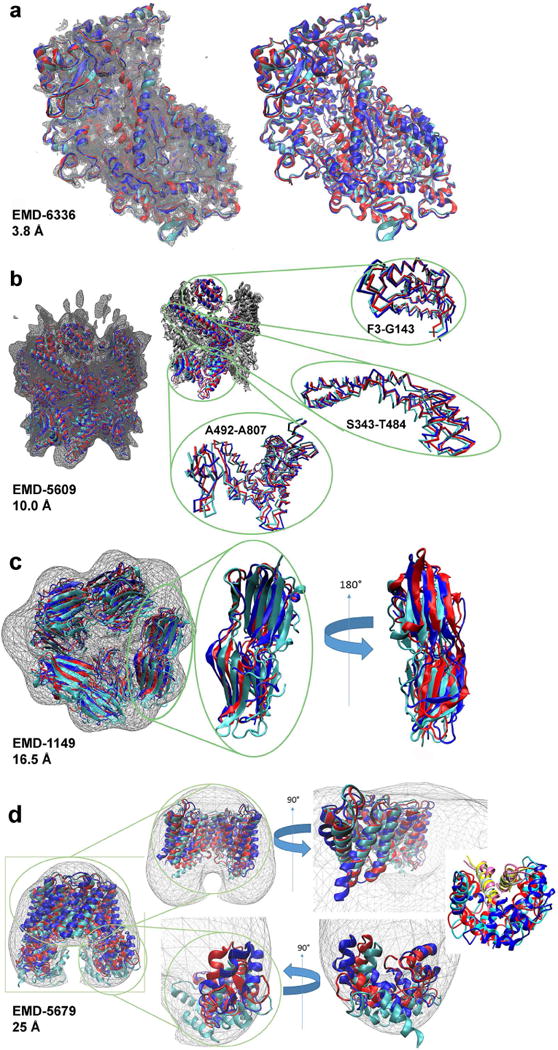
An increasing number of biomolecular structures are solved by electron microscopy (EM). However, the quality of structure models determined from EM maps vary substantially. To understand to what extent structure models are supported by information embedded in EM maps, we used two computational structure refinement methods to examine how much structures can be refined using a dataset of 49 maps with accompanying structure models. The extent of structure modification as well as the disagreement between refinement models produced by the two computational methods scaled inversely with the global and the local map resolutions. A general quantitative estimation of deviations of structures for particular map resolutions are provided. Our results indicate that the observed discrepancy between the deposited map and the refined models is due to the lack of structural information present in EM maps and thus these annotations must be used with caution for further applications.
Keywords: EMDB; computational modelling; cryo-EM; electron microscopy; model refinement; protein structure modelling; protein tertiary structure; structure biology; structure optimization.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Figures


Similar articles
-
Computational methods for constructing protein structure models from 3D electron microscopy maps.J Struct Biol. 2013 Oct;184(1):93-102. doi: 10.1016/j.jsb.2013.06.008. Epub 2013 Jun 21. J Struct Biol. 2013. PMID: 23796504 Free PMC article.
-
VESPER: global and local cryo-EM map alignment using local density vectors.Nat Commun. 2021 Apr 7;12(1):2090. doi: 10.1038/s41467-021-22401-y. Nat Commun. 2021. PMID: 33828103 Free PMC article.
-
Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta.Elife. 2016 Sep 26;5:e17219. doi: 10.7554/eLife.17219. Elife. 2016. PMID: 27669148 Free PMC article.
-
Evolution of standardization and dissemination of cryo-EM structures and data jointly by the community, PDB, and EMDB.J Biol Chem. 2021 Jan-Jun;296:100560. doi: 10.1016/j.jbc.2021.100560. Epub 2021 Mar 18. J Biol Chem. 2021. PMID: 33744287 Free PMC article. Review.
-
Refinement of Atomic Structures Against cryo-EM Maps.Methods Enzymol. 2016;579:277-305. doi: 10.1016/bs.mie.2016.05.033. Epub 2016 Jun 24. Methods Enzymol. 2016. PMID: 27572731 Review.
Cited by
-
CryoEM-based hybrid modeling approaches for structure determination.Curr Opin Microbiol. 2018 Jun;43:14-23. doi: 10.1016/j.mib.2017.10.002. Epub 2017 Nov 4. Curr Opin Microbiol. 2018. PMID: 29107896 Free PMC article. Review.
-
Macromolecular refinement of X-ray and cryoelectron microscopy structures with Phenix/OPLS3e for improved structure and ligand quality.Structure. 2021 Aug 5;29(8):913-921.e4. doi: 10.1016/j.str.2021.03.011. Epub 2021 Apr 5. Structure. 2021. PMID: 33823127 Free PMC article.
-
Constructing atomic structural models into cryo-EM densities using molecular dynamics - Pros and cons.J Struct Biol. 2018 Nov;204(2):319-328. doi: 10.1016/j.jsb.2018.08.003. Epub 2018 Aug 7. J Struct Biol. 2018. PMID: 30092279 Free PMC article.
-
In situ architecture of the ER-mitochondria encounter structure.Nature. 2023 Jun;618(7963):188-192. doi: 10.1038/s41586-023-06050-3. Epub 2023 May 10. Nature. 2023. PMID: 37165187 Free PMC article.
-
Simulation-Based Methods for Model Building and Refinement in Cryoelectron Microscopy.J Chem Inf Model. 2020 May 26;60(5):2470-2483. doi: 10.1021/acs.jcim.0c00087. Epub 2020 Mar 31. J Chem Inf Model. 2020. PMID: 32202798 Free PMC article. Review.
References
-
- Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868. - PubMed
-
- Ceulemans H, Russell RB. Fast fitting of atomic structures to low-resolution electron density maps by surface overlap maximization. Journal of molecular biology. 2004;338:783–793. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
