

To address surface reaction network complexity using scaling relations machine learning and DFT calculations
- PMID: 28262694
- PMCID: PMC5343483
- DOI: 10.1038/ncomms14621
To address surface reaction network complexity using scaling relations machine learning and DFT calculations
Abstract
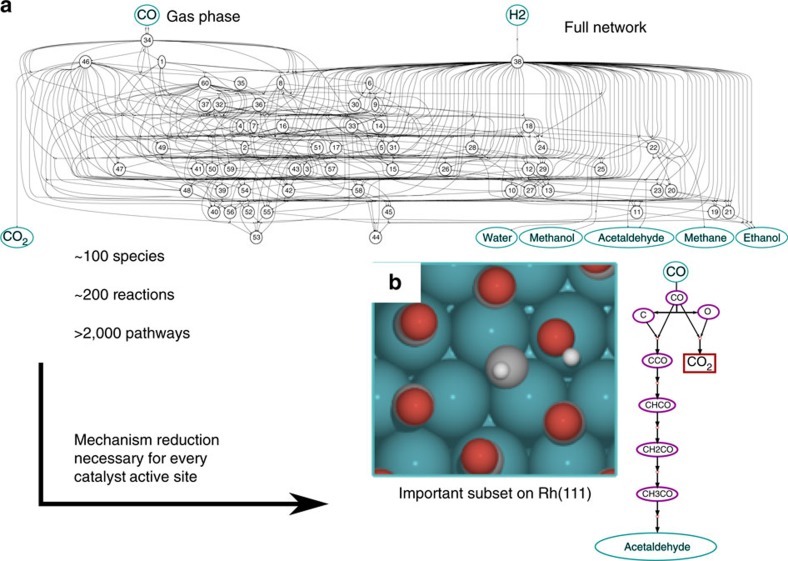
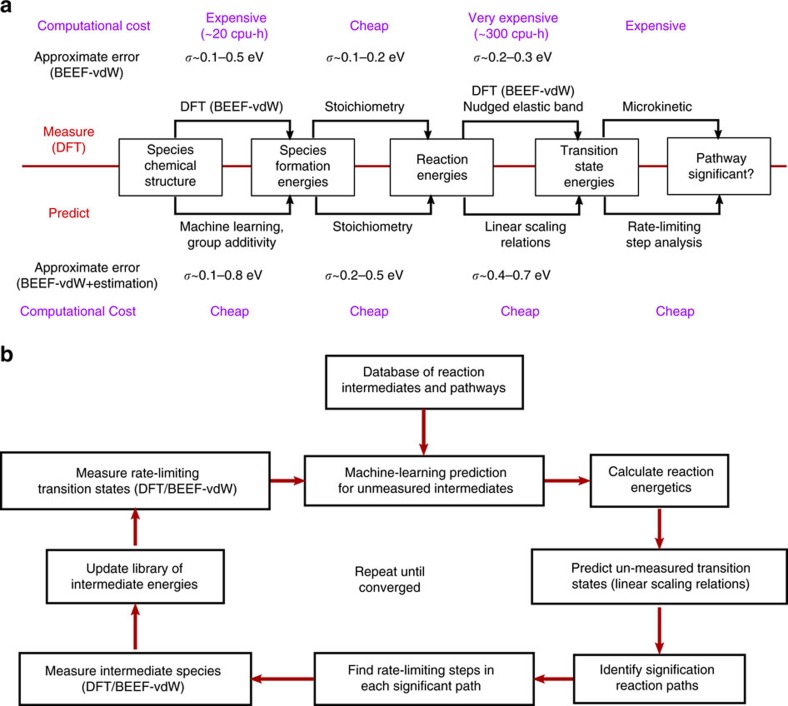
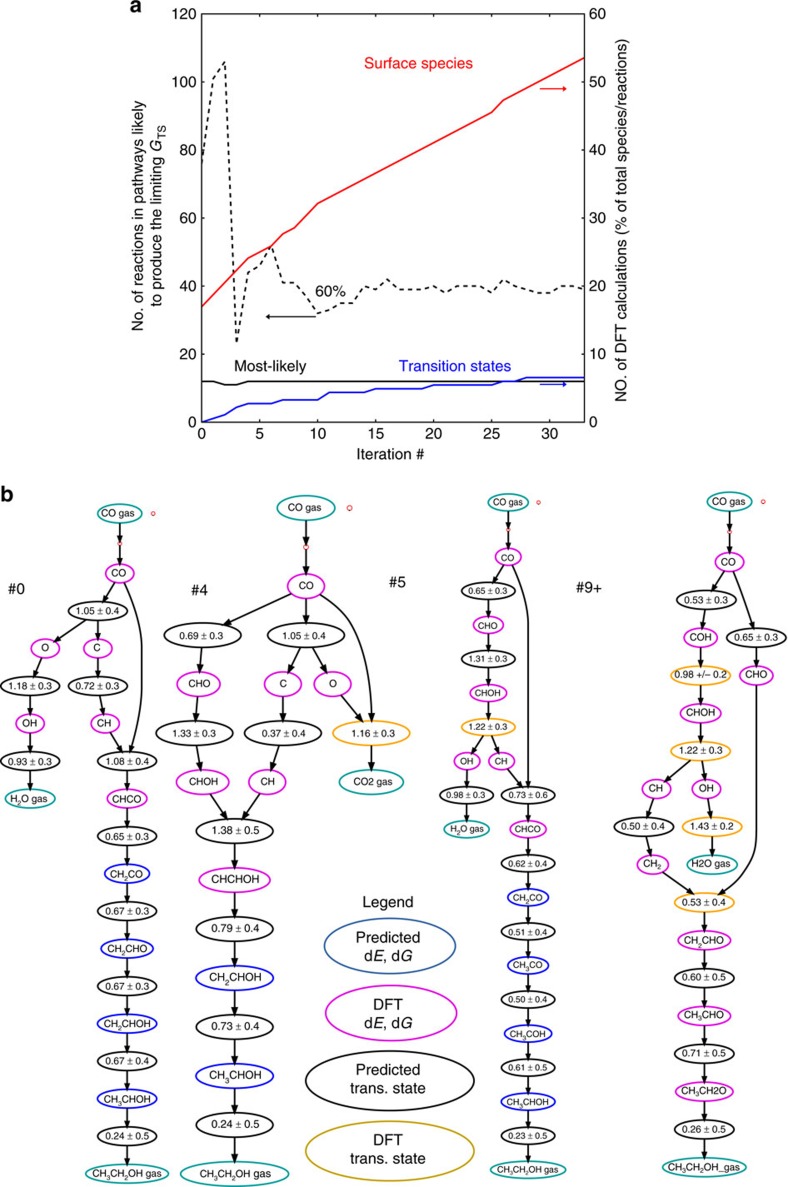
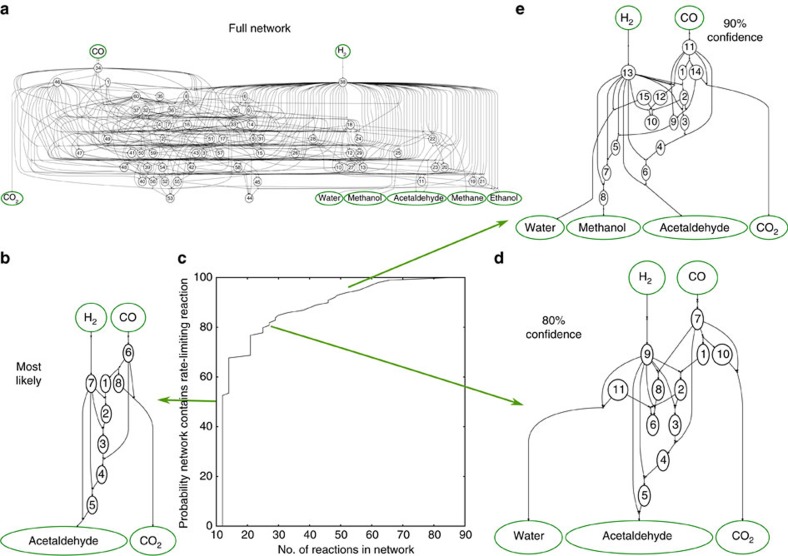
Surface reaction networks involving hydrocarbons exhibit enormous complexity with thousands of species and reactions for all but the very simplest of chemistries. We present a framework for optimization under uncertainty for heterogeneous catalysis reaction networks using surrogate models that are trained on the fly. The surrogate model is constructed by teaching a Gaussian process adsorption energies based on group additivity fingerprints, combined with transition-state scaling relations and a simple classifier for determining the rate-limiting step. The surrogate model is iteratively used to predict the most important reaction step to be calculated explicitly with computationally demanding electronic structure theory. Applying these methods to the reaction of syngas on rhodium(111), we identify the most likely reaction mechanism. Propagating uncertainty throughout this process yields the likelihood that the final mechanism is complete given measurements on only a subset of the entire network and uncertainty in the underlying density functional theory calculations.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Gao C. W., Allen J. W., Green W. H. & West R. H. Reaction mechanism generator: automatic construction of chemical kinetic mechanisms. Comput. Phys. Commun. 203, 212–255 (2016).
-
- Nørskov J. K., Bligaard T., Rossmeisl J. & Christensen C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009). - PubMed
-
- Salciccioli M., Chen Y. & Vlachos D. G. Density Functional Theory-derived group additivity and linear scaling methods for prediction of oxygenate stability on metal catalysts: adsorption of open-ring alcohol and polyol dehydrogenation intermediates on Pt-based metals. J. Phys. Chem. C 114, 20155–20166 (2010).
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
