

Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia
- PMID: 28263186
- PMCID: PMC5373861
- DOI: 10.1172/JCI90193
Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia
Abstract
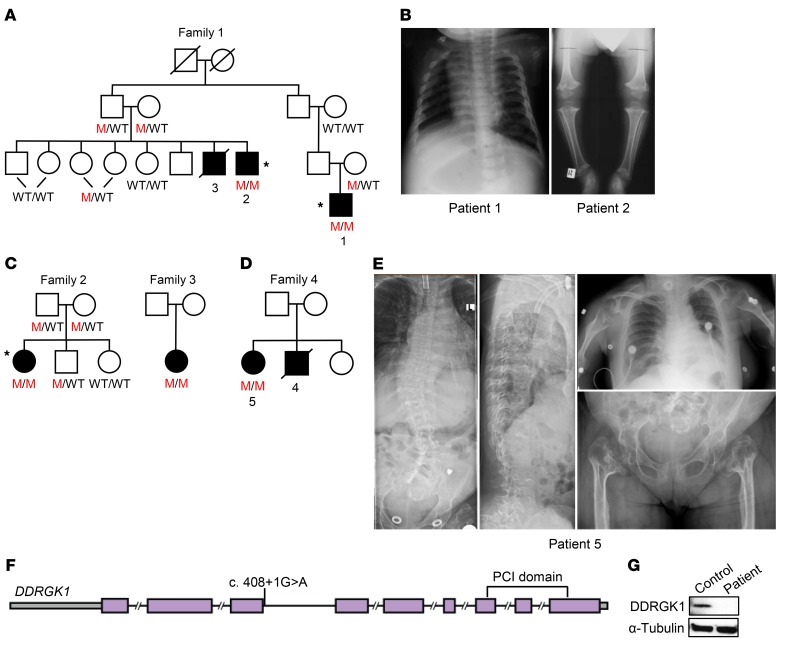
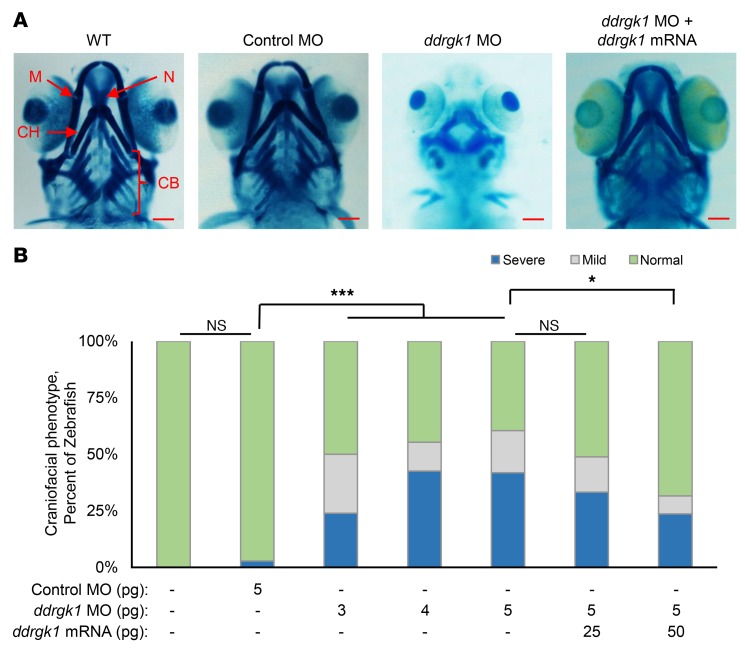
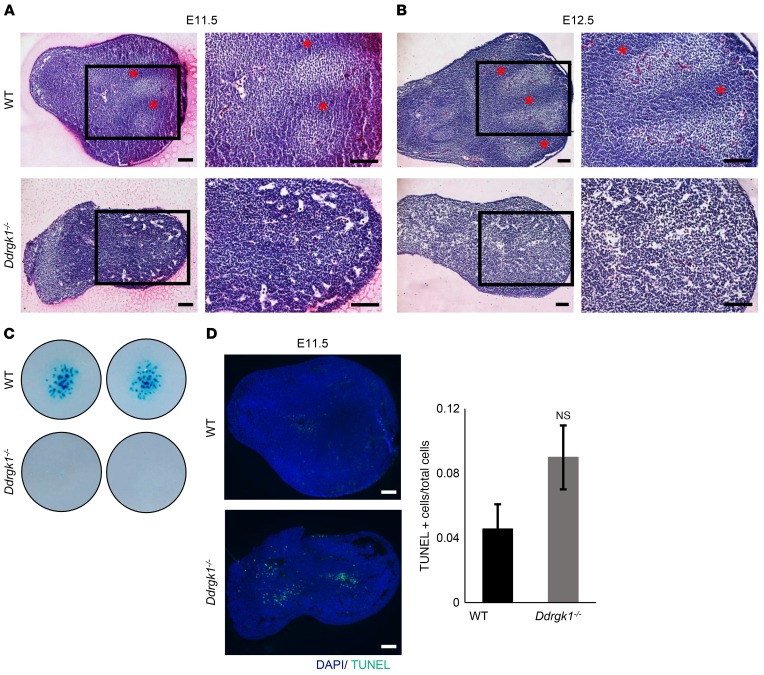
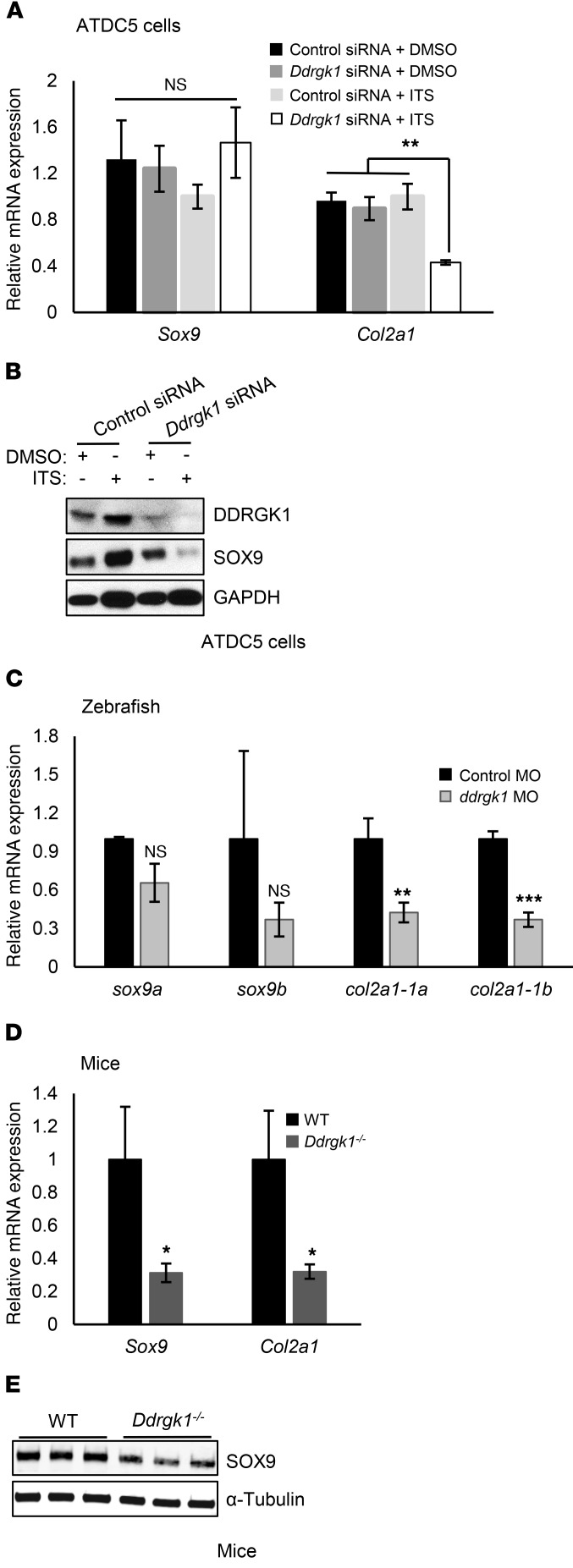
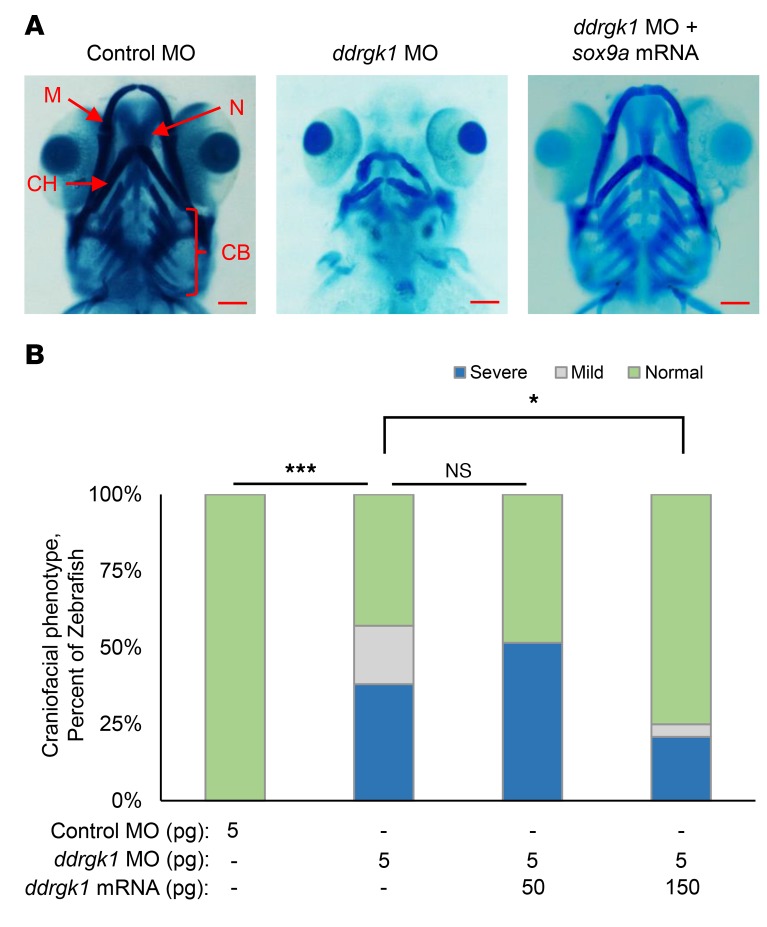
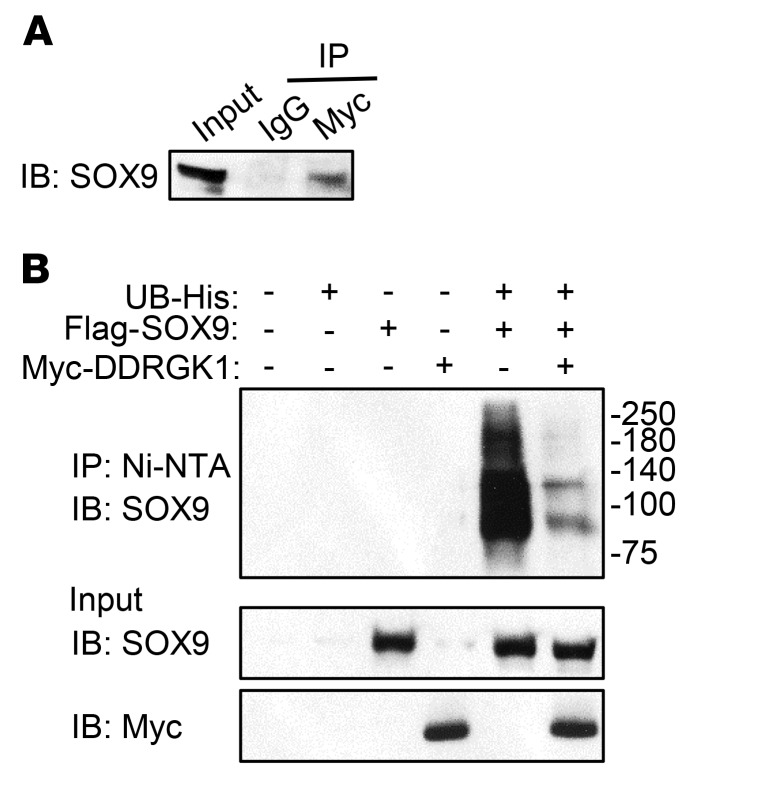
Shohat-type spondyloepimetaphyseal dysplasia (SEMD) is a skeletal dysplasia that affects cartilage development. Similar skeletal disorders, such as spondyloepiphyseal dysplasias, are linked to mutations in type II collagen (COL2A1), but the causative gene in SEMD is not known. Here, we have performed whole-exome sequencing to identify a recurrent homozygous c.408+1G>A donor splice site loss-of-function mutation in DDRGK domain containing 1 (DDRGK1) in 4 families affected by SEMD. In zebrafish, ddrgk1 deficiency disrupted craniofacial cartilage development and led to decreased levels of the chondrogenic master transcription factor sox9 and its downstream target, col2a1. Overexpression of sox9 rescued the zebrafish chondrogenic and craniofacial phenotype generated by ddrgk1 knockdown, thus identifying DDRGK1 as a regulator of SOX9. Consistent with these results, Ddrgk1-/- mice displayed delayed limb bud chondrogenic condensation, decreased SOX9 protein expression and Col2a1 transcript levels, and increased apoptosis. Furthermore, we determined that DDRGK1 can directly bind to SOX9 to inhibit its ubiquitination and proteasomal degradation. Taken together, these data indicate that loss of DDRGK1 decreases SOX9 expression and causes a human skeletal dysplasia, identifying a mechanism that regulates chondrogenesis via modulation of SOX9 ubiquitination.
Conflict of interest statement
Figures
References
-
- Spranger J, Winterpacht A, Zabel B. The type II collagenopathies: a spectrum of chondrodysplasias. Eur J Pediatr. 1994;153(2):56–65. - PubMed
-
- Kuivaniemi H, Tromp G, Prockop DJ. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat. 1997;9(4):300–315. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
