

Deep-sea vent phage DNA polymerase specifically initiates DNA synthesis in the absence of primers
- PMID: 28265063
- PMCID: PMC5373334
- DOI: 10.1073/pnas.1700280114
Deep-sea vent phage DNA polymerase specifically initiates DNA synthesis in the absence of primers
Abstract
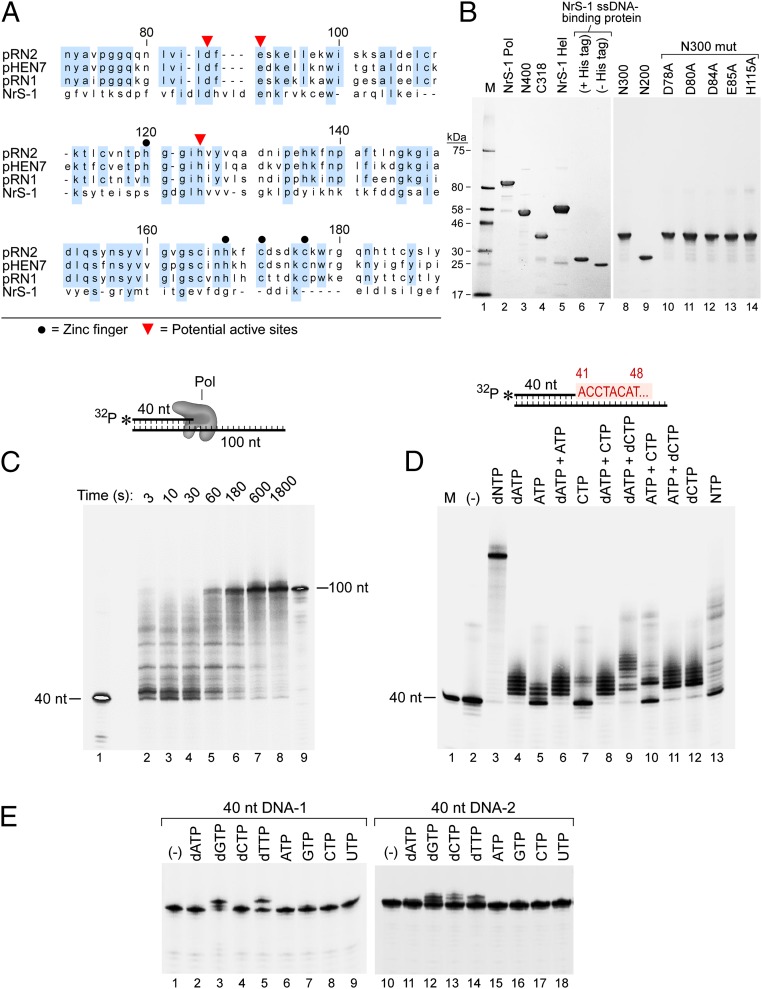
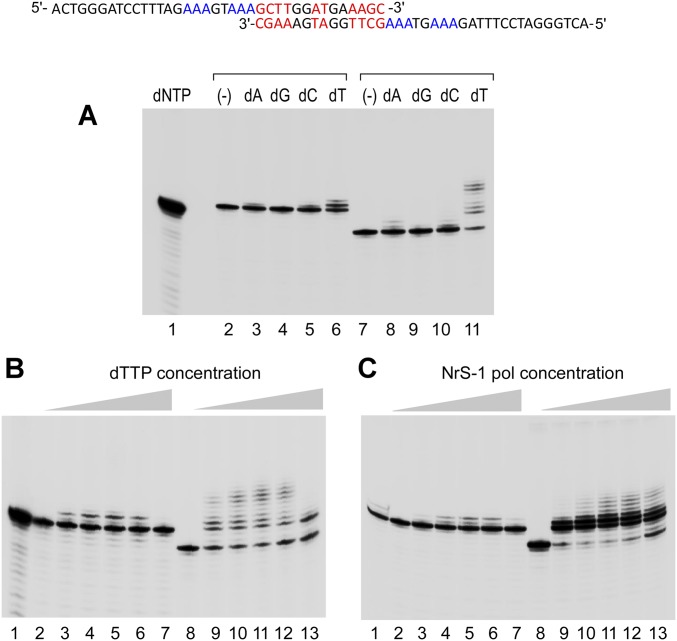
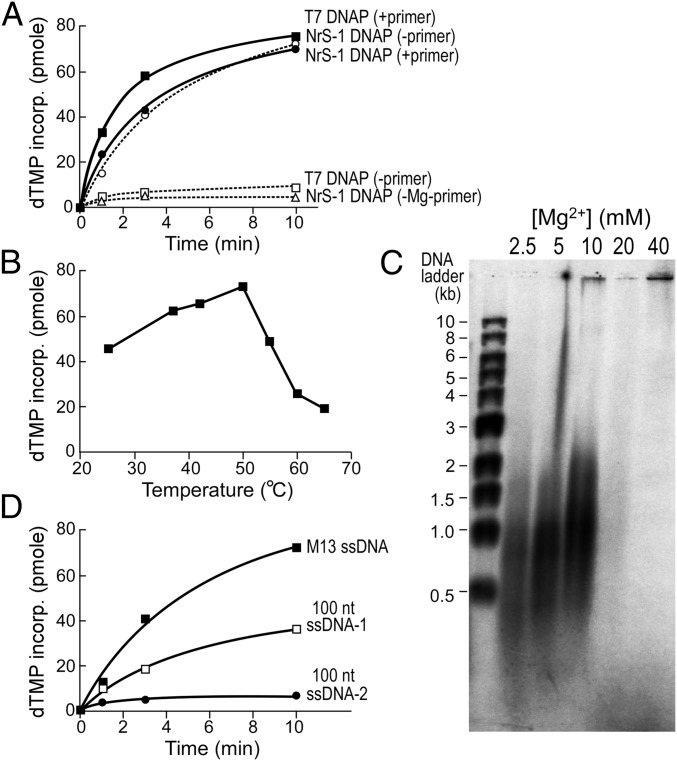
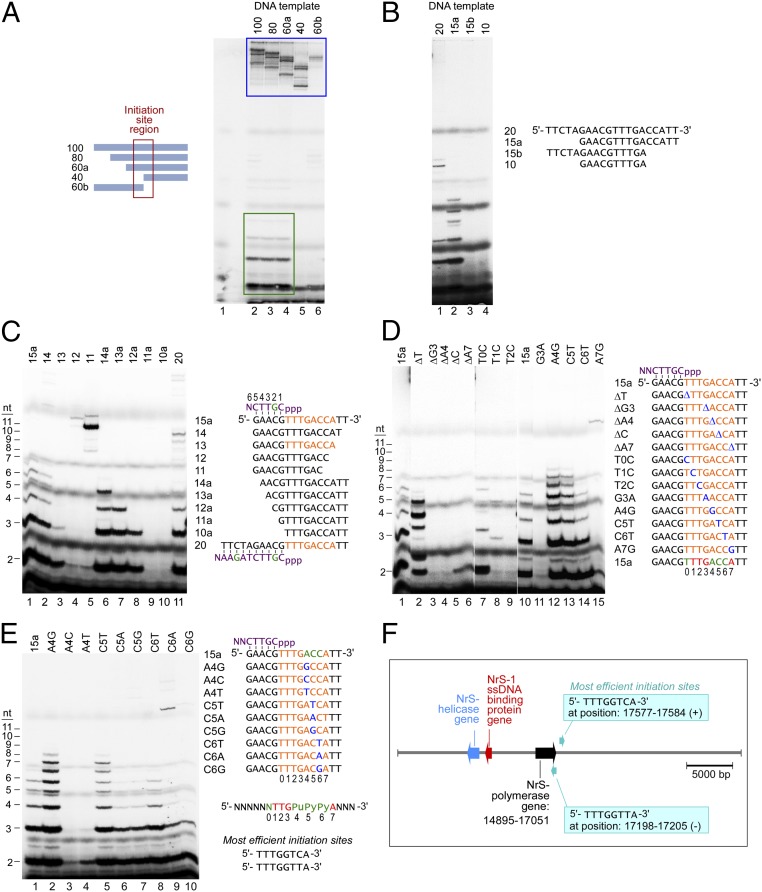
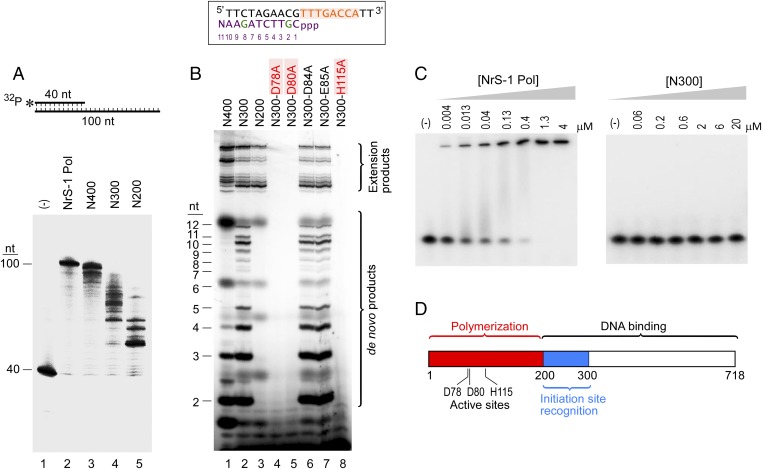
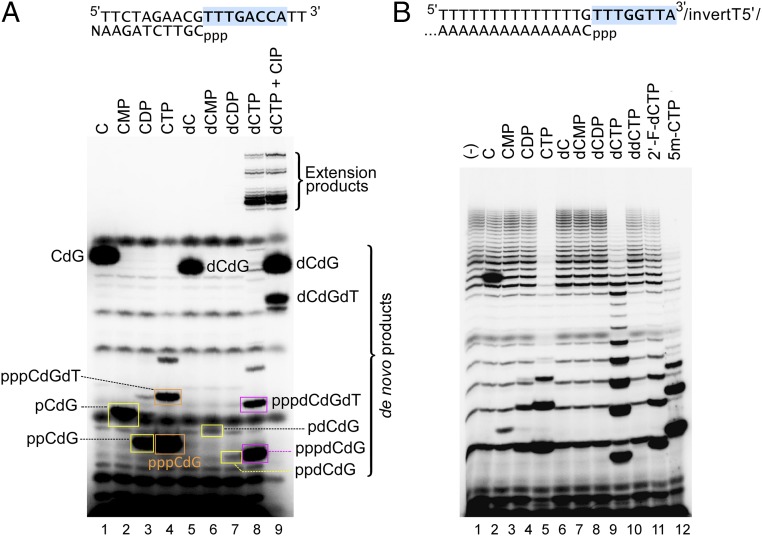
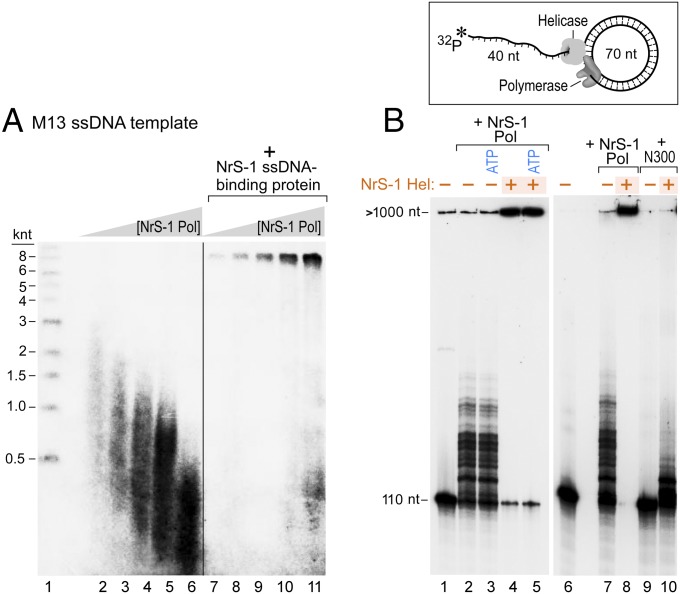
A DNA polymerase is encoded by the deep-sea vent phage NrS-1. NrS-1 has a unique genome organization containing genes that are predicted to encode a helicase and a single-stranded DNA (ssDNA)-binding protein. The gene for an unknown protein shares weak homology with the bifunctional primase-polymerases (prim-pols) from archaeal plasmids but is missing the zinc-binding domain typically found in primases. We show that this gene product has efficient DNA polymerase activity and is processive in DNA synthesis in the presence of the NrS-1 helicase and ssDNA-binding protein. Remarkably, this NrS-1 DNA polymerase initiates DNA synthesis from a specific template DNA sequence in the absence of any primer. The de novo DNA polymerase activity resides in the N-terminal domain of the protein, whereas the C-terminal domain enhances DNA binding.
Keywords: NrS-1; helicase; primase; prim–pol; ssDNA-binding protein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Frick DN, Richardson CC. DNA primases. Annu Rev Biochem. 2001;70:39–80. - PubMed
-
- Cheetham GM, Steitz TA. Insights into transcription: Structure and function of single-subunit DNA-dependent RNA polymerases. Curr Opin Struct Biol. 2000;10(1):117–123. - PubMed
-
- Liu L, et al. The archaeal DNA primase: Biochemical characterization of the p41-p46 complex from Pyrococcus furiosus. J Biol Chem. 2001;276(48):45484–45490. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
