

Precision medicine driven by cancer systems biology
- PMID: 28265786
- PMCID: PMC5385204
- DOI: 10.1007/s10555-017-9662-4
Precision medicine driven by cancer systems biology
Abstract
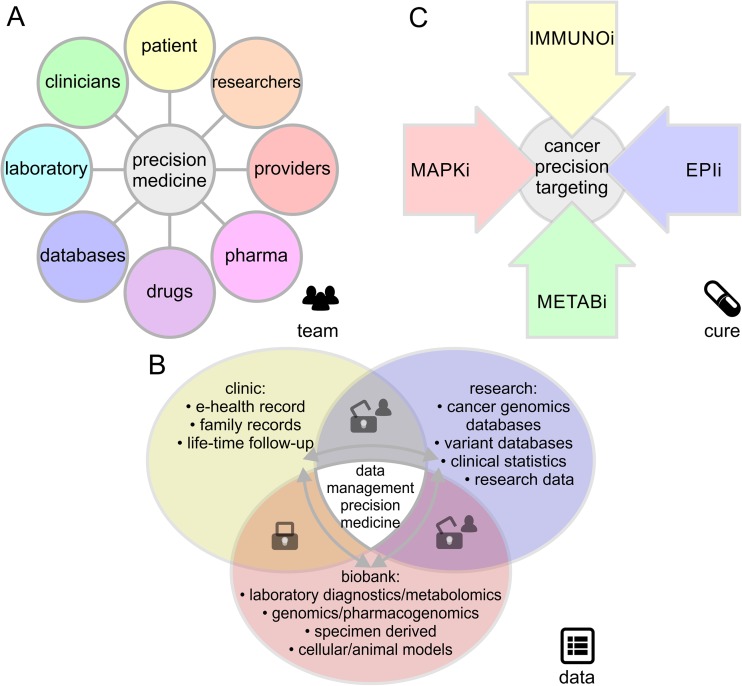
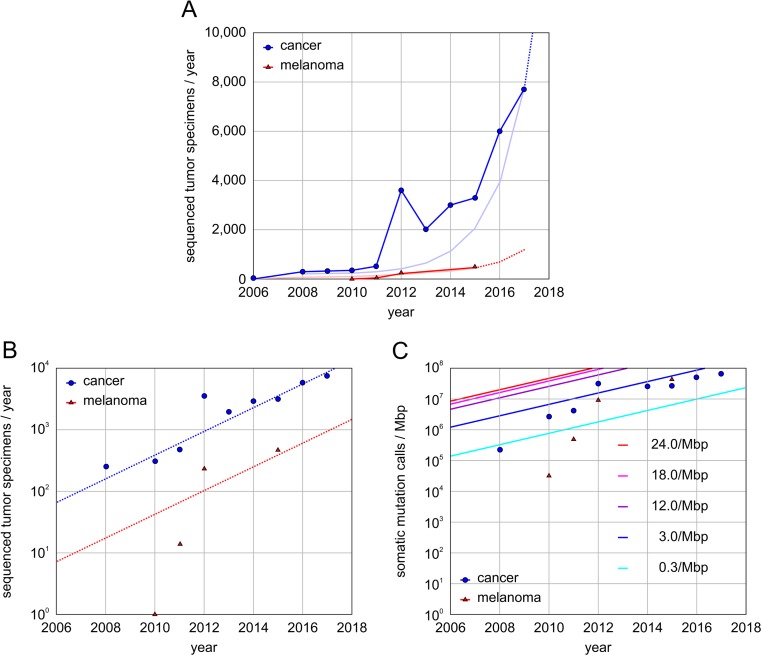
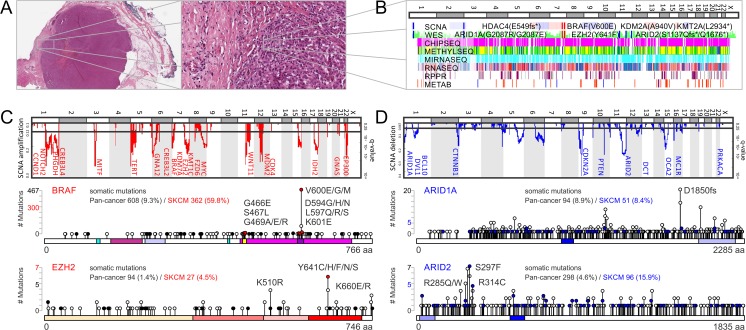
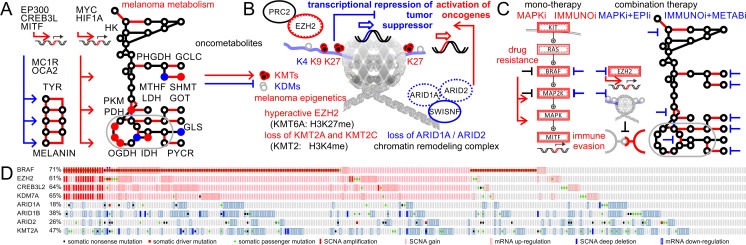
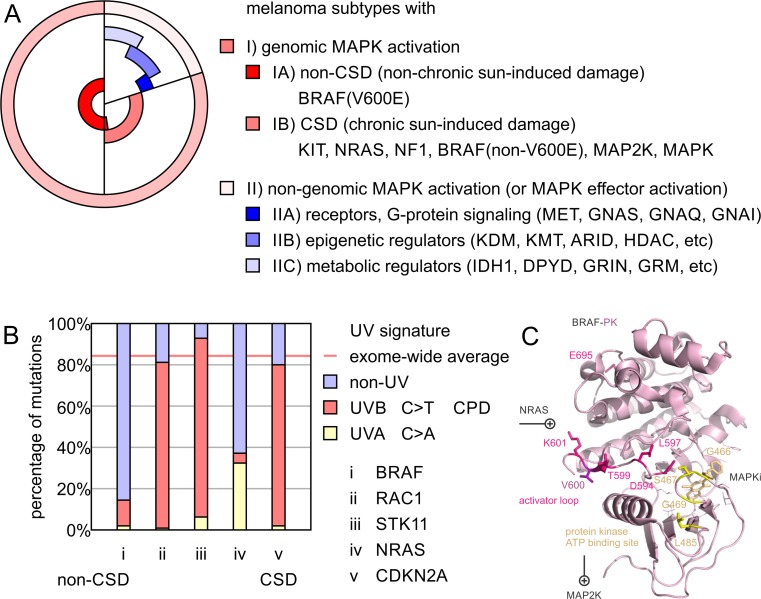
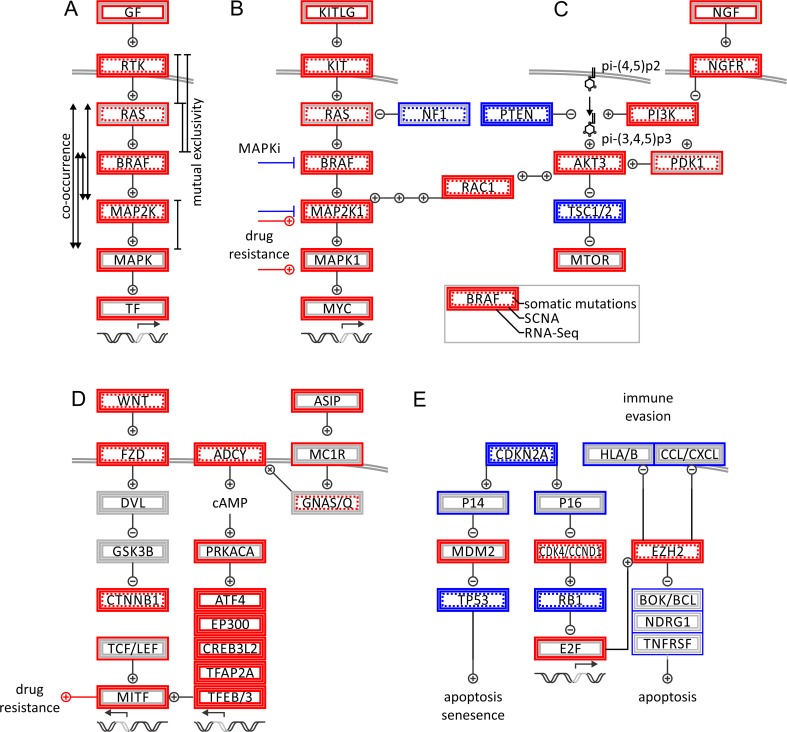
Molecular insights from genome and systems biology are influencing how cancer is diagnosed and treated. We critically evaluate big data challenges in precision medicine. The melanoma research community has identified distinct subtypes involving chronic sun-induced damage and the mitogen-activated protein kinase driver pathway. In addition, despite low mutation burden, non-genomic mitogen-activated protein kinase melanoma drivers are found in membrane receptors, metabolism, or epigenetic signaling with the ability to bypass central mitogen-activated protein kinase molecules and activating a similar program of mitogenic effectors. Mutation hotspots, structural modeling, UV signature, and genomic as well as non-genomic mechanisms of disease initiation and progression are taken into consideration to identify resistance mutations and novel drug targets. A comprehensive precision medicine profile of a malignant melanoma patient illustrates future rational drug targeting strategies. Network analysis emphasizes an important role of epigenetic and metabolic master regulators in oncogenesis. Co-occurrence of driver mutations in signaling, metabolic, and epigenetic factors highlights how cumulative alterations of our genomes and epigenomes progressively lead to uncontrolled cell proliferation. Precision insights have the ability to identify independent molecular pathways suitable for drug targeting. Synergistic treatment combinations of orthogonal modalities including immunotherapy, mitogen-activated protein kinase inhibitors, epigenetic inhibitors, and metabolic inhibitors have the potential to overcome immune evasion, side effects, and drug resistance.
Keywords: ARID1A; ARID2; BRAF; Big data; CSD; CTLA4; Cancer metabolism; Cancer systems biology; Combination therapy; Driver; EZH2; Epigenomics; Immunotherapy; MEK; Melanoma; Neoantigen; Oncometabolite; PD1; PDL1; PRC2; Personalized medicine; Precision medicine; SCNA; SWI/SNF; Subtype.
Conflict of interest statement
Conflict of interest
There is no conflict of interest.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
