

Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population
- PMID: 28266639
- PMCID: PMC5339803
- DOI: 10.1038/srep44185
Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population
Abstract
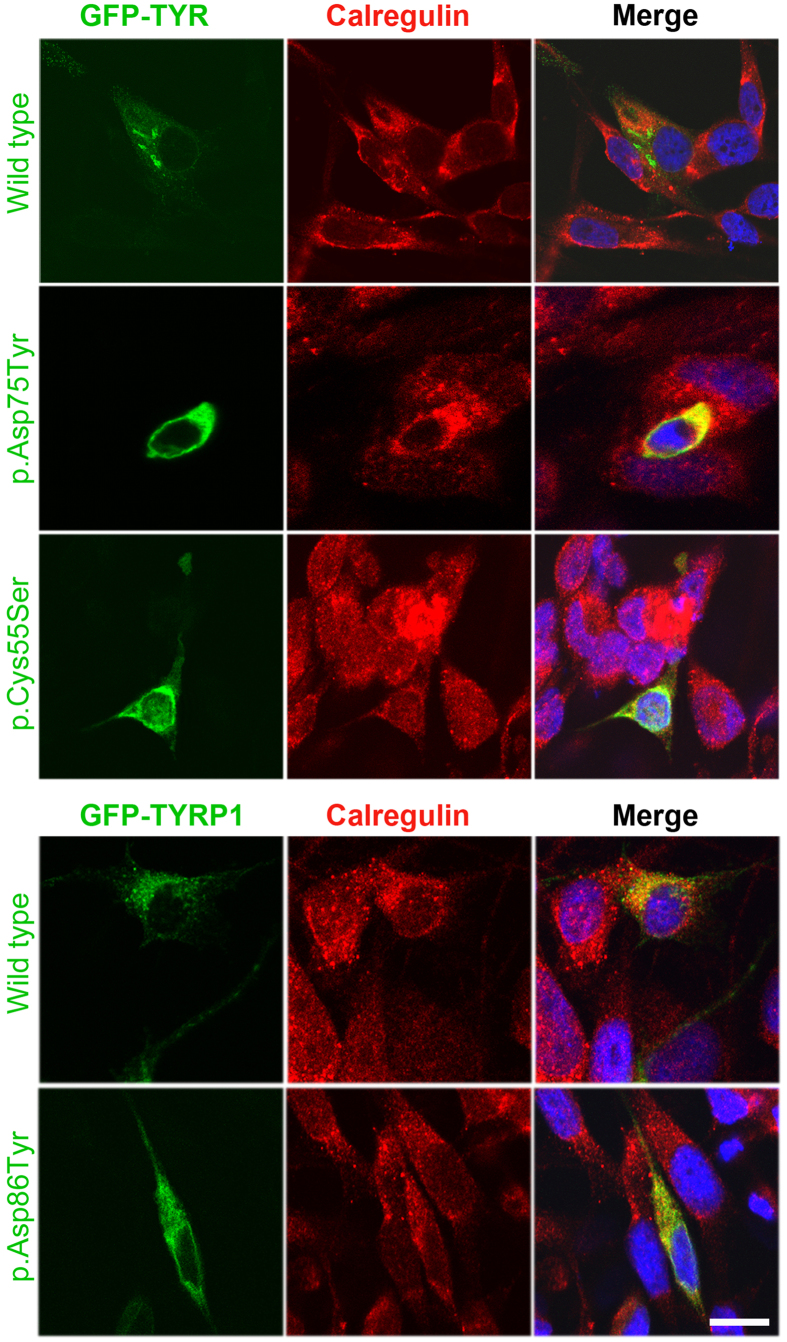
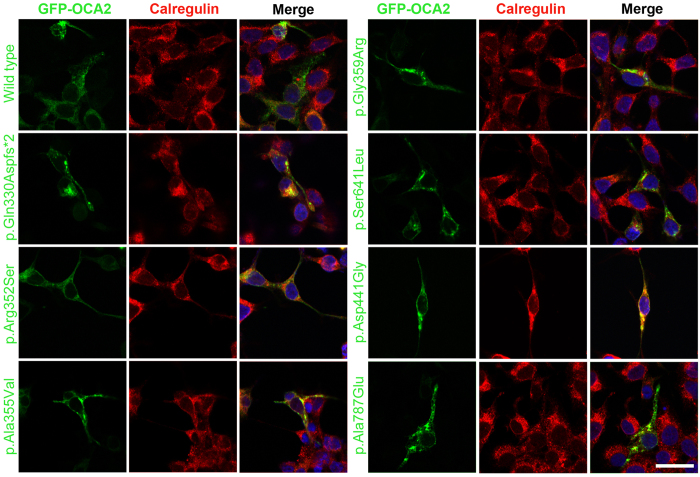
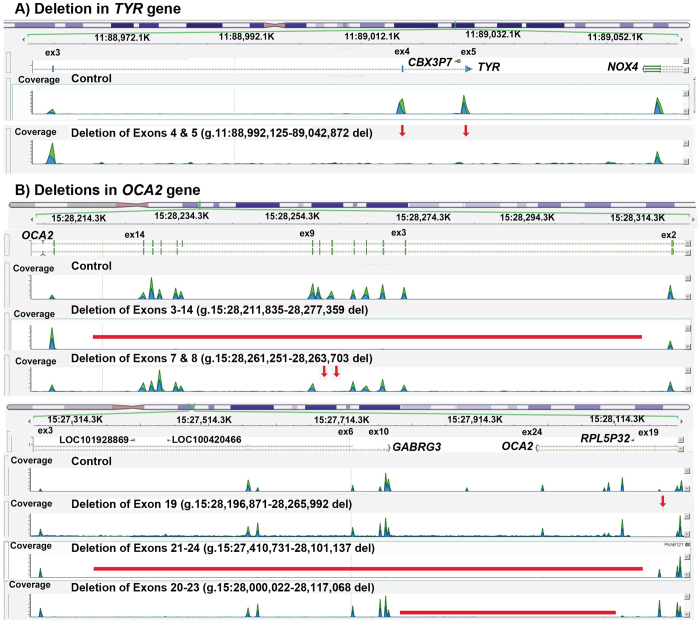
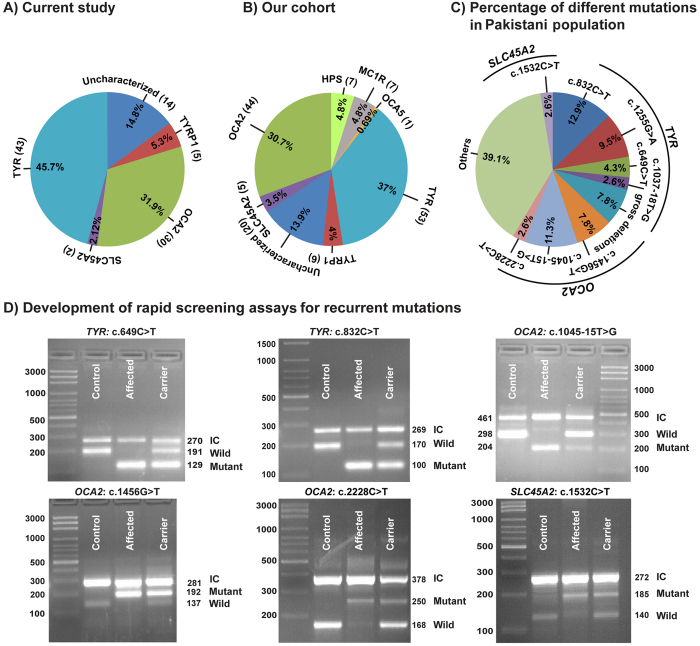
Nonsyndromic oculocutaneous Albinism (nsOCA) is clinically characterized by the loss of pigmentation in the skin, hair, and iris. OCA is amongst the most common causes of vision impairment in children. To date, pathogenic variants in six genes have been identified in individuals with nsOCA. Here, we determined the identities, frequencies, and clinical consequences of OCA alleles in 94 previously unreported Pakistani families. Combination of Sanger and Exome sequencing revealed 38 alleles, including 22 novel variants, segregating with nsOCA phenotype in 80 families. Variants of TYR and OCA2 genes were the most common cause of nsOCA, occurring in 43 and 30 families, respectively. Twenty-two novel variants include nine missense, four splice site, two non-sense, one insertion and six gross deletions. In vitro studies revealed retention of OCA proteins harboring novel missense alleles in the endoplasmic reticulum (ER) of transfected cells. Exon-trapping assays with constructs containing splice site alleles revealed errors in splicing. As eight alleles account for approximately 56% (95% CI: 46.52-65.24%) of nsOCA cases, primarily enrolled from Punjab province of Pakistan, hierarchical strategies for variant detection would be feasible and cost-efficient genetic tests for OCA in families with similar origin. Thus, we developed Tetra-primer ARMS assays for rapid, reliable, reproducible and economical screening of most of these common alleles.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

References
-
- Spritz R. A., Chiang P. W., Oiso N. & Alkhateeb A. Human and mouse disorders of pigmentation. Curr Opin Genet Dev 13, 284–289 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
