

Quirks of Error Estimation in Cross-Linking/Mass Spectrometry
- PMID: 28267312
- PMCID: PMC5423704
- DOI: 10.1021/acs.analchem.6b03745
Quirks of Error Estimation in Cross-Linking/Mass Spectrometry
Abstract
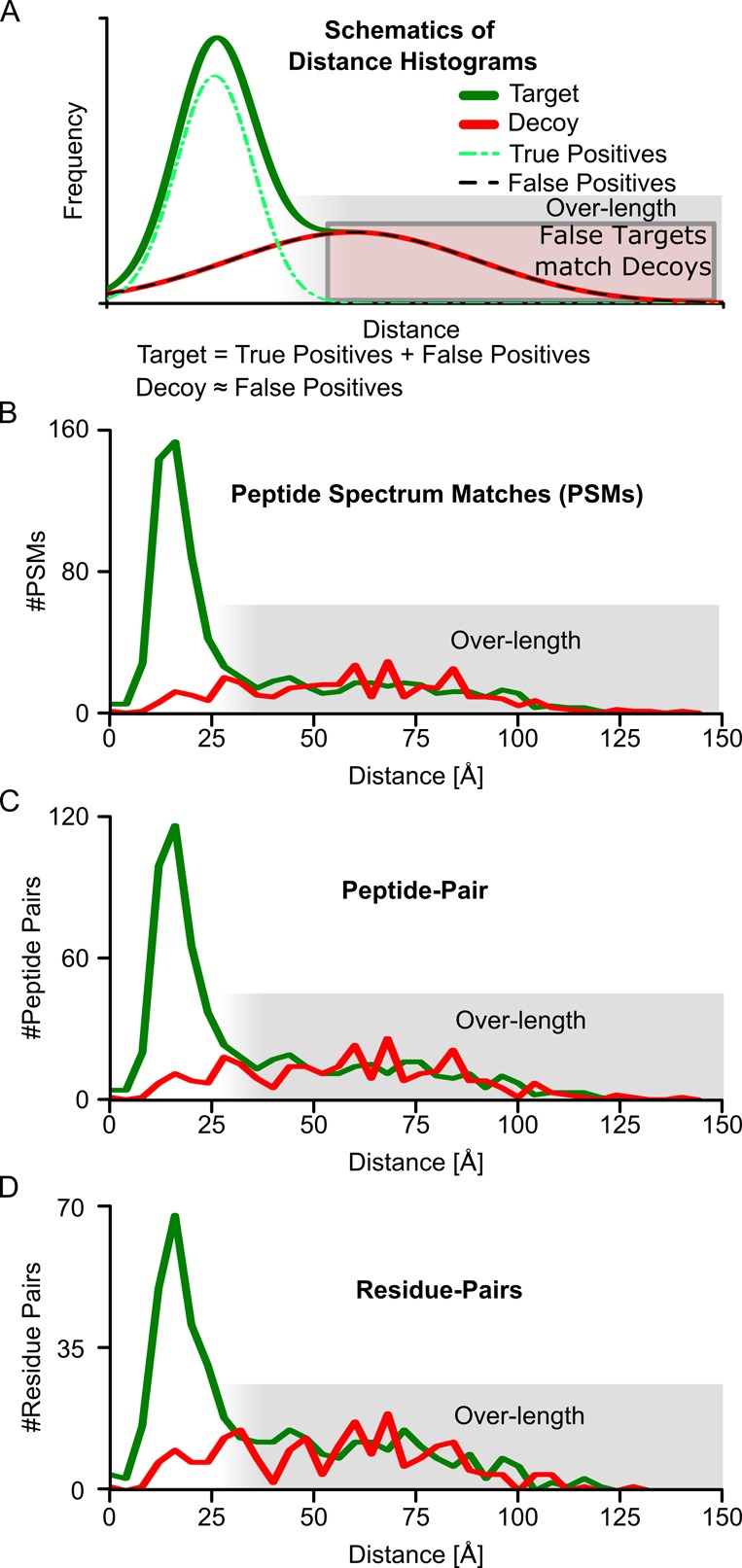
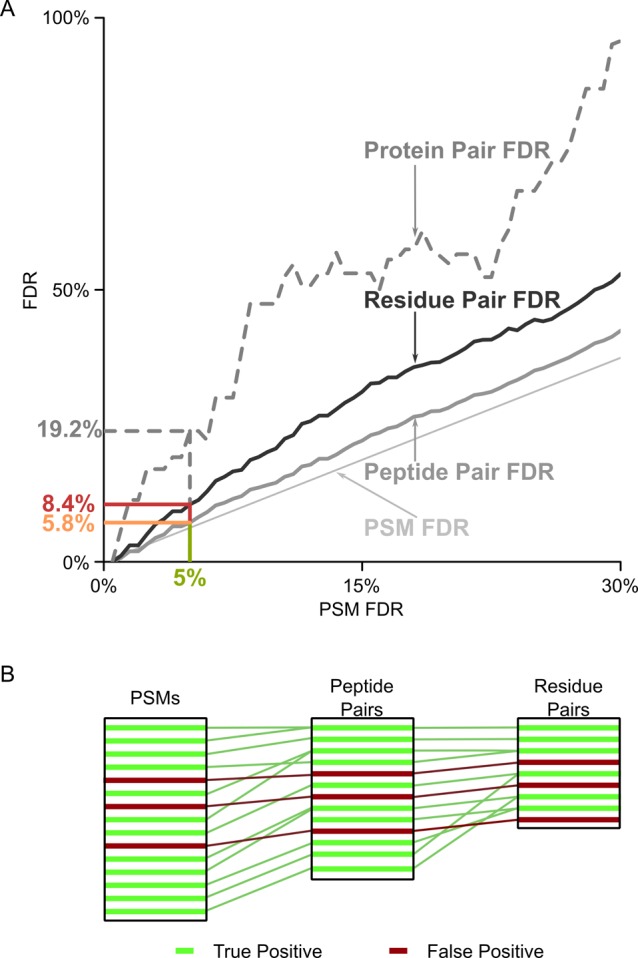
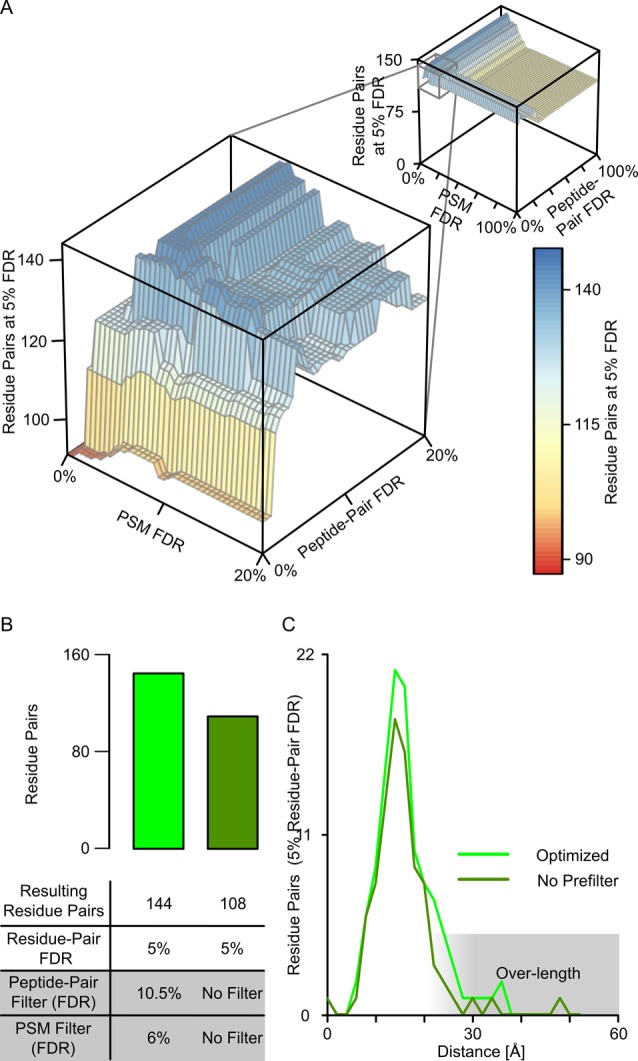
Cross-linking/mass spectrometry is an increasingly popular approach to obtain structural information on proteins and their complexes in solution. However, methods for error assessment are under current development. We note that false-discovery rates can be estimated at different points during data analysis, and are most relevant for residue or protein pairs. Missing this point led in our example analysis to an actual 8.4% error when 5% error was targeted. In addition, prefiltering of peptide-spectrum matches and of identified peptide pairs substantially improved results. In our example, this prefiltering increased the number of residue pairs (5% FDR) by 33% (n = 108 to n = 144). This number improvement did not come at the expense of reduced accuracy as the added data agreed with an available crystal structure. We provide an open-source tool, xiFDR ( https://github.com/rappsilberlab/xiFDR ), that implements our observations for routine application. Data are available via ProteomeXchange with identifier PXD004749.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
