

Prototype foamy virus elicits complete autophagy involving the ER stress-related UPR pathway
- PMID: 28270144
- PMCID: PMC5341167
- DOI: 10.1186/s12977-017-0341-x
Prototype foamy virus elicits complete autophagy involving the ER stress-related UPR pathway
Abstract
Background: Prototype foamy virus (PFV) is a member of the Spumaretrovirinae subfamily of retroviruses, which maintains lifelong latent infection while being nonpathogenic to their natural hosts. Autophagy is a cell-programmed mechanism that plays a pivotal role in controlling homeostasis and defense against exotic pathogens. However, whether autophagy is the mechanism for host defense in PFV infection has not been investigated.
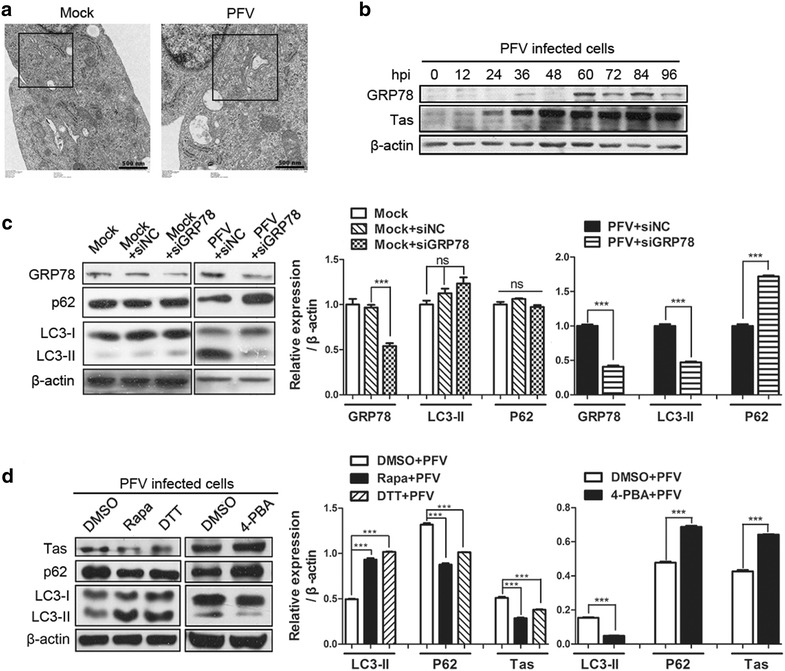
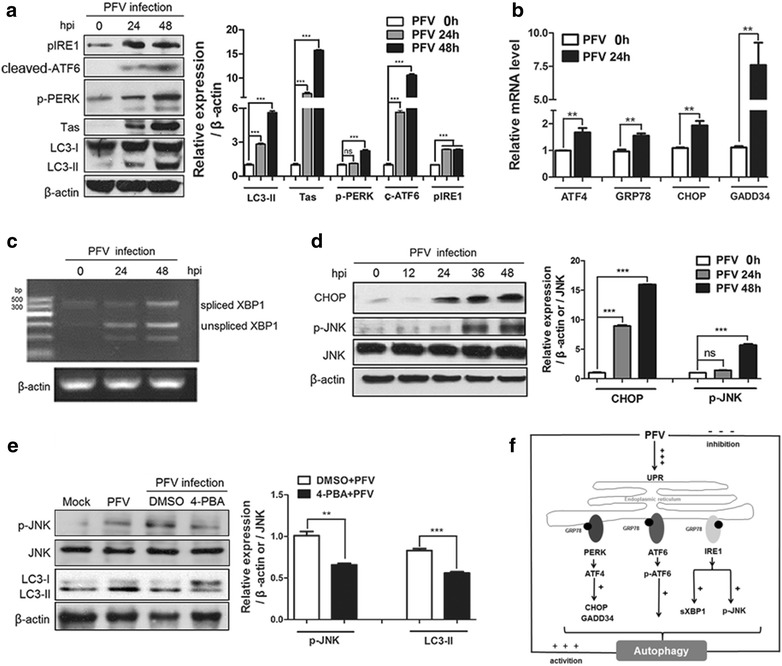
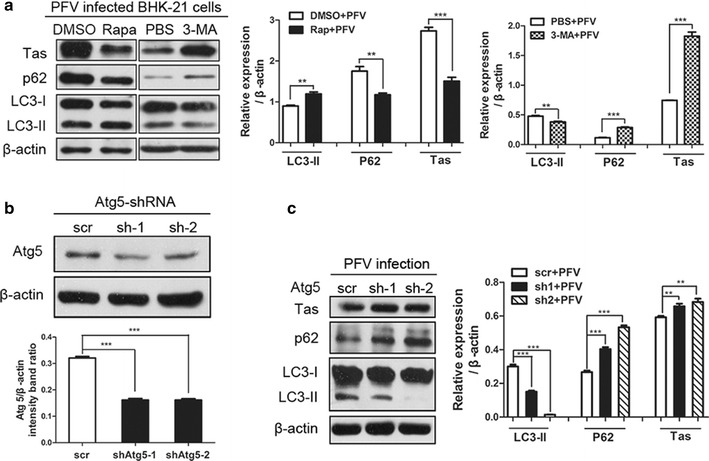
Findings: Our results revealed that PFV infection induced the accumulation of autophagosomes and triggered complete autophagic flux in BHK-21 cells. PFV infection also altered endoplasmic reticulum (ER) homeostasis. The PERK, IRE1 and ATF6 pathways, all of which are components of the ER stress-related unfolded protein response (UPR), were activated in PFV-infected cells. In addition, accelerating autophagy suppressed PFV replication, and inhibition of autophagy promoted viral replication.
Conclusions: Our data indicate that PFV infection can induce complete autophagy through activating the ER stress-related UPR pathway in BHK-21 cells. In turn, autophagy negatively regulates PFV replication.
Keywords: Autophagy; ER stress; PFV; Viral replication.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
