

Molecular Population Genetics
- PMID: 28270526
- PMCID: PMC5340319
- DOI: 10.1534/genetics.116.196493
Molecular Population Genetics
Erratum in
-
Molecular Population Genetics.Genetics. 2019 Oct;213(2):721-722. doi: 10.1534/genetics.119.302623. Genetics. 2019. PMID: 31594909 Free PMC article. No abstract available.
Abstract
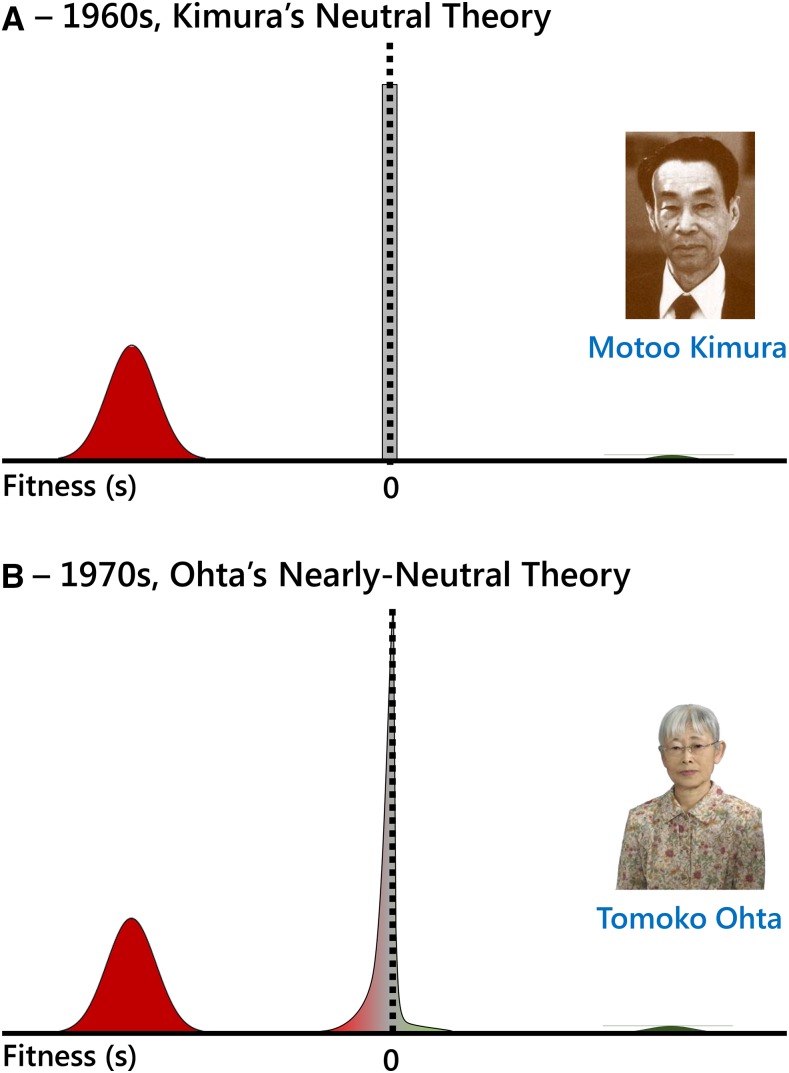
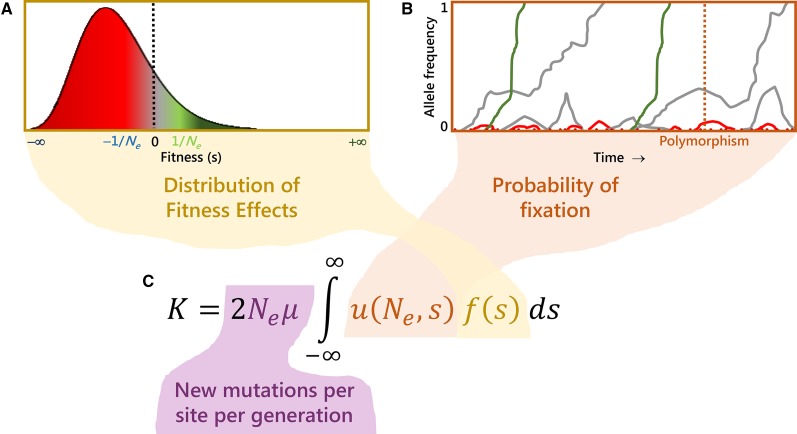
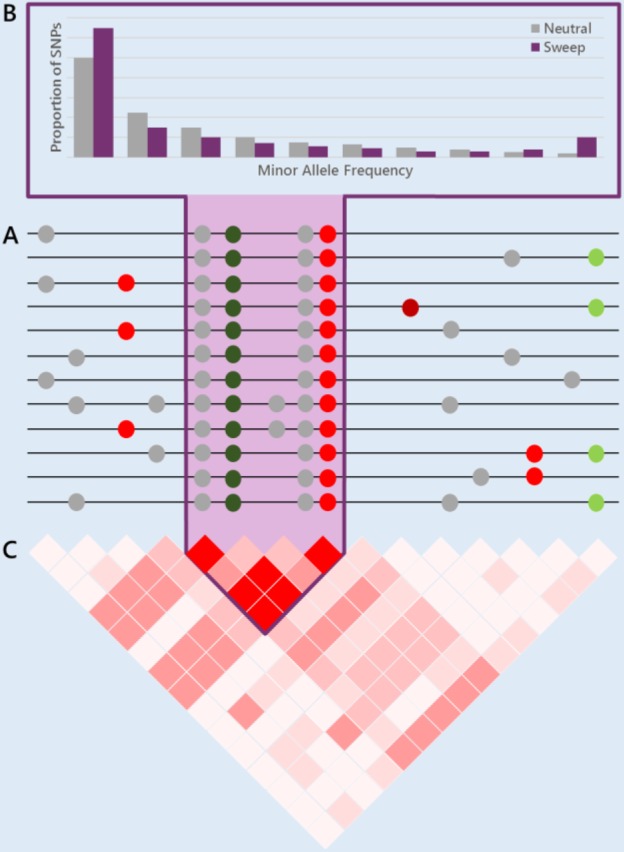
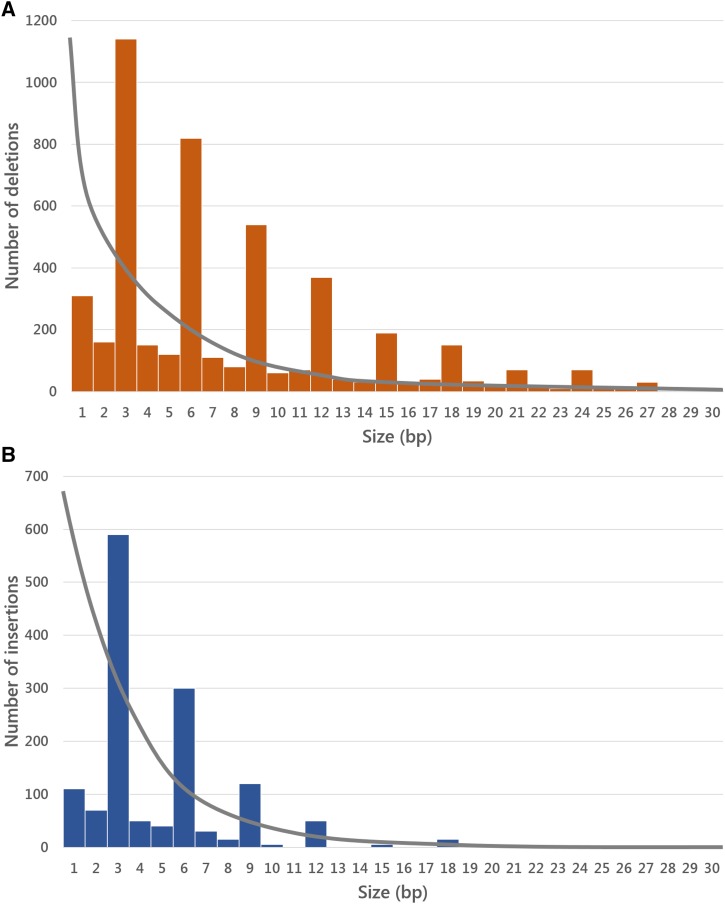
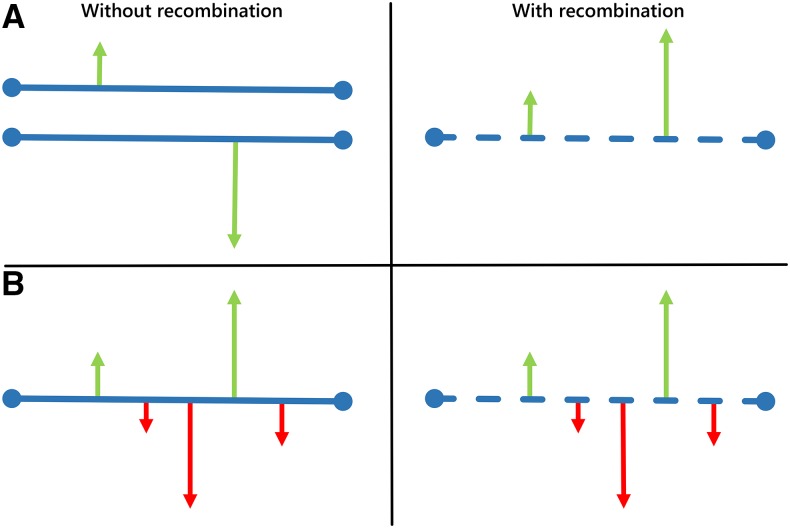
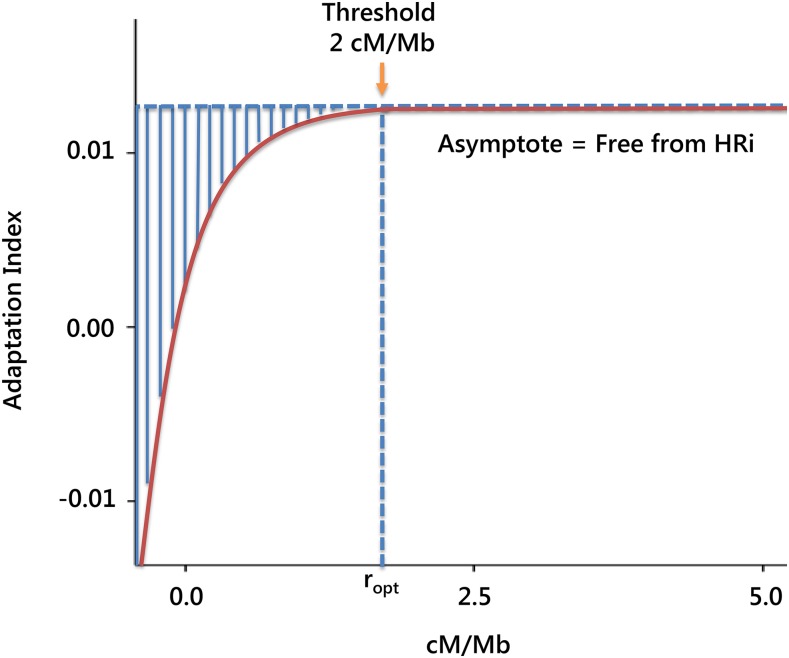
Molecular population genetics aims to explain genetic variation and molecular evolution from population genetics principles. The field was born 50 years ago with the first measures of genetic variation in allozyme loci, continued with the nucleotide sequencing era, and is currently in the era of population genomics. During this period, molecular population genetics has been revolutionized by progress in data acquisition and theoretical developments. The conceptual elegance of the neutral theory of molecular evolution or the footprint carved by natural selection on the patterns of genetic variation are two examples of the vast number of inspiring findings of population genetics research. Since the inception of the field, Drosophila has been the prominent model species: molecular variation in populations was first described in Drosophila and most of the population genetics hypotheses were tested in Drosophila species. In this review, we describe the main concepts, methods, and landmarks of molecular population genetics, using the Drosophila model as a reference. We describe the different genetic data sets made available by advances in molecular technologies, and the theoretical developments fostered by these data. Finally, we review the results and new insights provided by the population genomics approach, and conclude by enumerating challenges and new lines of inquiry posed by increasingly large population scale sequence data.
Keywords: Drosophila; FlyBook; Hill–Robertson interference; distribution of fitness effects; genetic draft; linked selection; molecular population genetics; neutral theory; population genomics; population multi-omics.
Copyright © 2017 Casillas and Barbadilla.
Figures
References
-
- Adams M. D., Celniker S. E., Holt R. A., Evans C. A., Gocayne J. D., et al. , 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
