

Principles of Reconstructing the Subclonal Architecture of Cancers
- PMID: 28270531
- PMCID: PMC5538405
- DOI: 10.1101/cshperspect.a026625
Principles of Reconstructing the Subclonal Architecture of Cancers
Abstract
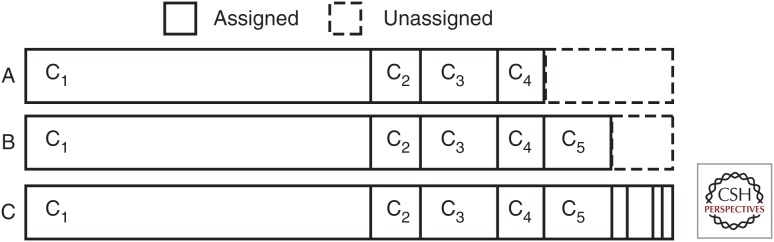
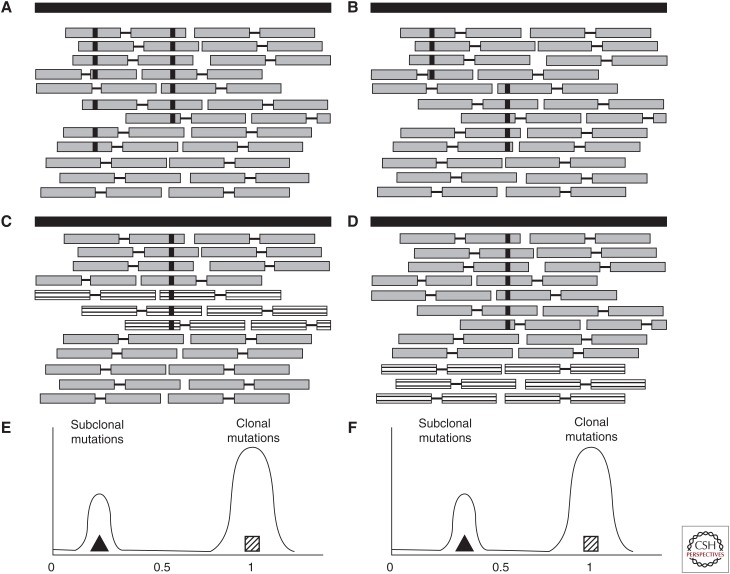
Most cancers evolve from a single founder cell through a series of clonal expansions that are driven by somatic mutations. These clonal expansions can lead to several coexisting subclones sharing subsets of mutations. Analysis of massively parallel sequencing data can infer a tumor's subclonal composition through the identification of populations of cells with shared mutations. We describe the principles that underlie subclonal reconstruction through single nucleotide variants (SNVs) or copy number alterations (CNAs) from bulk or single-cell sequencing. These principles include estimating the fraction of tumor cells for SNVs and CNAs, performing clustering of SNVs from single- and multisample cases, and single-cell sequencing. The application of subclonal reconstruction methods is providing key insights into tumor evolution, identifying subclonal driver mutations, patterns of parallel evolution and differences in mutational signatures between cellular populations, and characterizing the mechanisms of therapy resistance, spread, and metastasis.
Copyright © 2017 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources