

Genetics of Amyotrophic Lateral Sclerosis
- PMID: 28270533
- PMCID: PMC5932579
- DOI: 10.1101/cshperspect.a024125
Genetics of Amyotrophic Lateral Sclerosis
Abstract
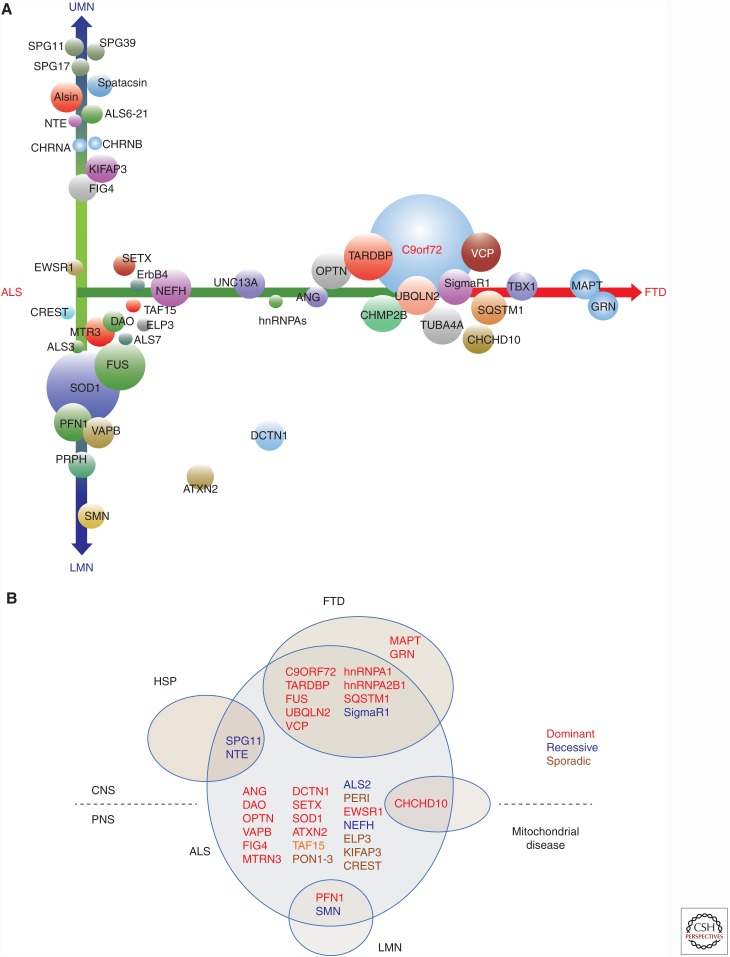
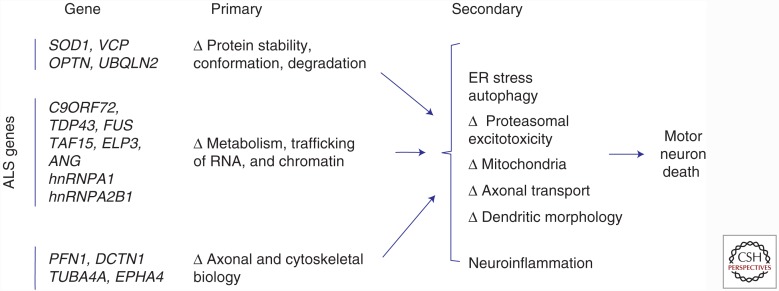
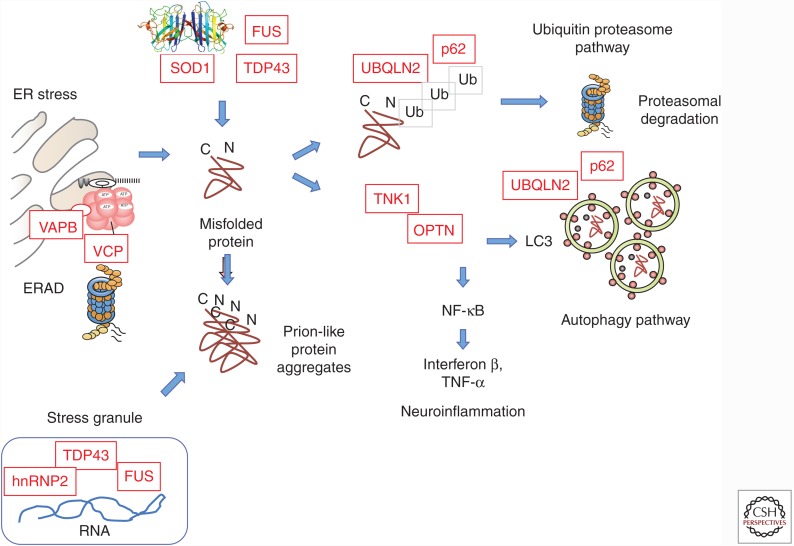
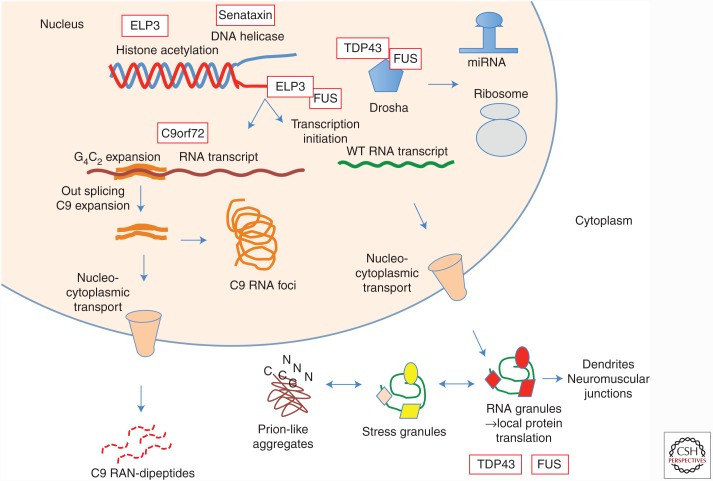
Amyotrophic lateral sclerosis (ALS) is a devastating, uniformly lethal degenerative disorder of motor neurons that overlaps clinically with frontotemporal dementia (FTD). Investigations of the 10% of ALS cases that are transmitted as dominant traits have revealed numerous gene mutations and variants that either cause these disorders or influence their clinical phenotype. The evolving understanding of the genetic architecture of ALS has illuminated broad themes in the molecular pathophysiology of both familial and sporadic ALS and FTD. These central themes encompass disturbances of protein homeostasis, alterations in the biology of RNA binding proteins, and defects in cytoskeletal dynamics, as well as numerous downstream pathophysiological events. Together, these findings from ALS genetics provide new insight into therapies that target genetically distinct subsets of ALS and FTD.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Abel O, Powell JF, Andersen PM, Al-Chalabi A. 2012. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat 33: 1345–1351. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous