

Acinar cell plasticity and development of pancreatic ductal adenocarcinoma
- PMID: 28270694
- PMCID: PMC6036907
- DOI: 10.1038/nrgastro.2017.12
Acinar cell plasticity and development of pancreatic ductal adenocarcinoma
Abstract
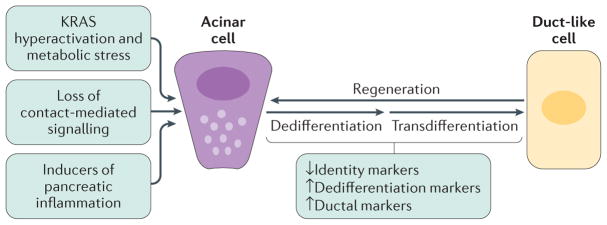
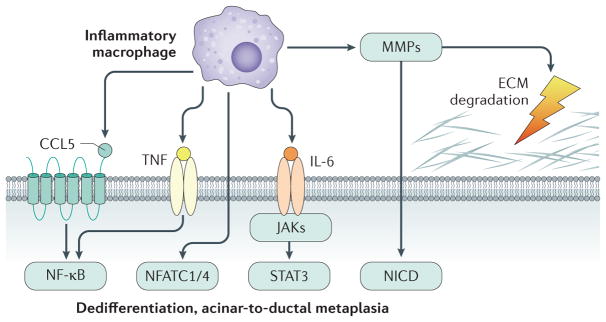
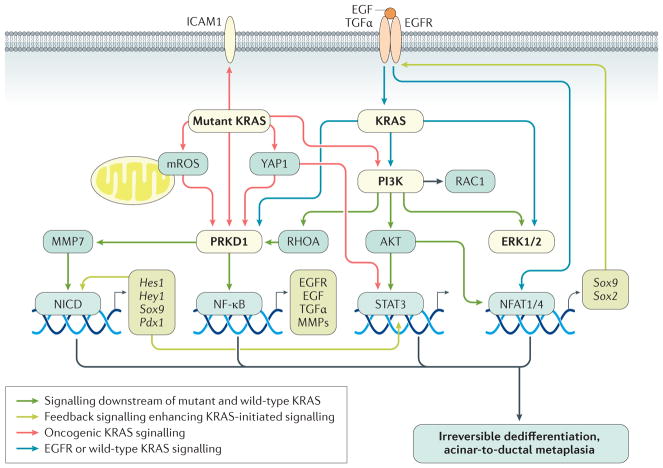
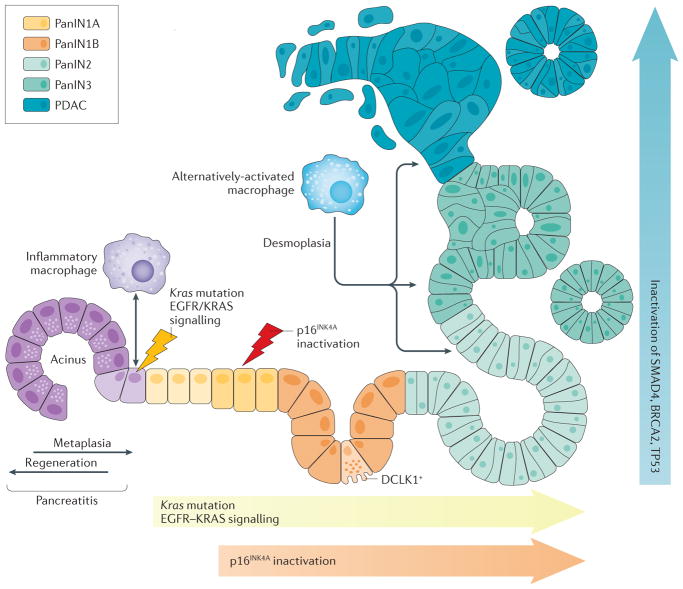
Acinar cells in the adult pancreas show high plasticity and can undergo transdifferentiation to a progenitor-like cell type with ductal characteristics. This process, termed acinar-to-ductal metaplasia (ADM), is an important feature facilitating pancreas regeneration after injury. Data from animal models show that cells that undergo ADM in response to oncogenic signalling are precursors for pancreatic intraepithelial neoplasia lesions, which can further progress to pancreatic ductal adenocarcinoma (PDAC). As human pancreatic adenocarcinoma is often diagnosed at a stage of metastatic disease, understanding the processes that lead to its initiation is important for the discovery of markers for early detection, as well as options that enable an early intervention. Here, the critical determinants of acinar cell plasticity are discussed, in addition to the intracellular and extracellular signalling events that drive acinar cell metaplasia and their contribution to development of PDAC.
Conflict of interest statement
The author declares no competing interests.
Figures
References
-
- Pinho AV, et al. Adult pancreatic acinar cells dedifferentiate to an embryonic progenitor phenotype with concomitant activation of a senescence programme that is present in chronic pancreatitis. Gut. 2011;60:958–966. - PubMed
-
- Rooman I, Real FX. Pancreatic ductal adenocarcinoma and acinar cells: a matter of differentiation and development? Gut. 2012;61:449–458. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
