

Complex multi-enhancer contacts captured by genome architecture mapping
- PMID: 28273065
- PMCID: PMC5366070
- DOI: 10.1038/nature21411
Complex multi-enhancer contacts captured by genome architecture mapping
Abstract
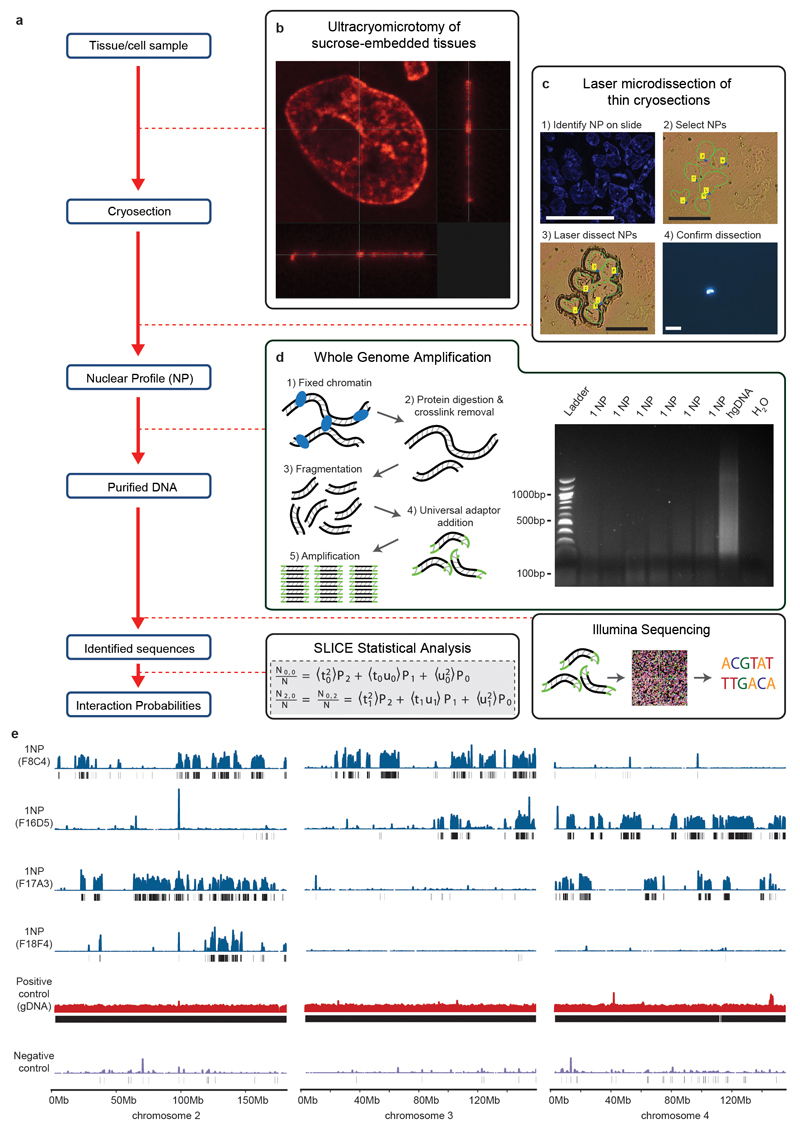
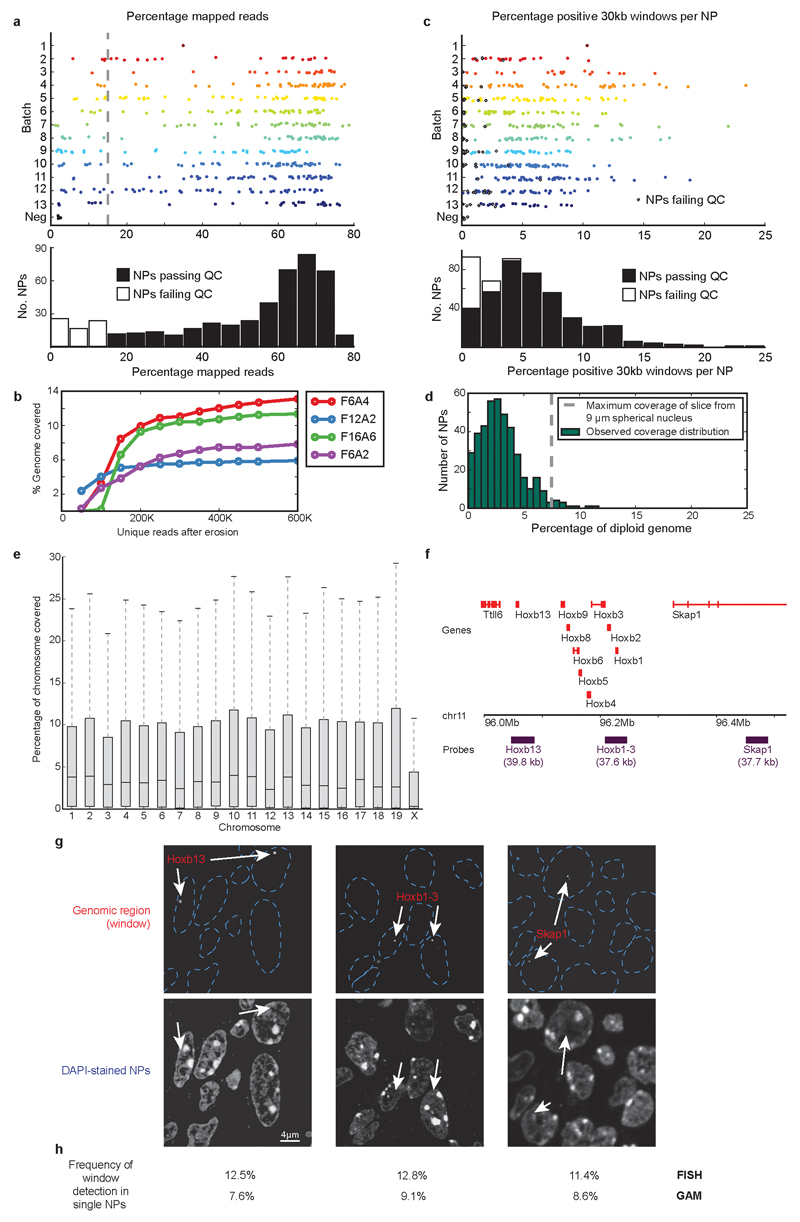
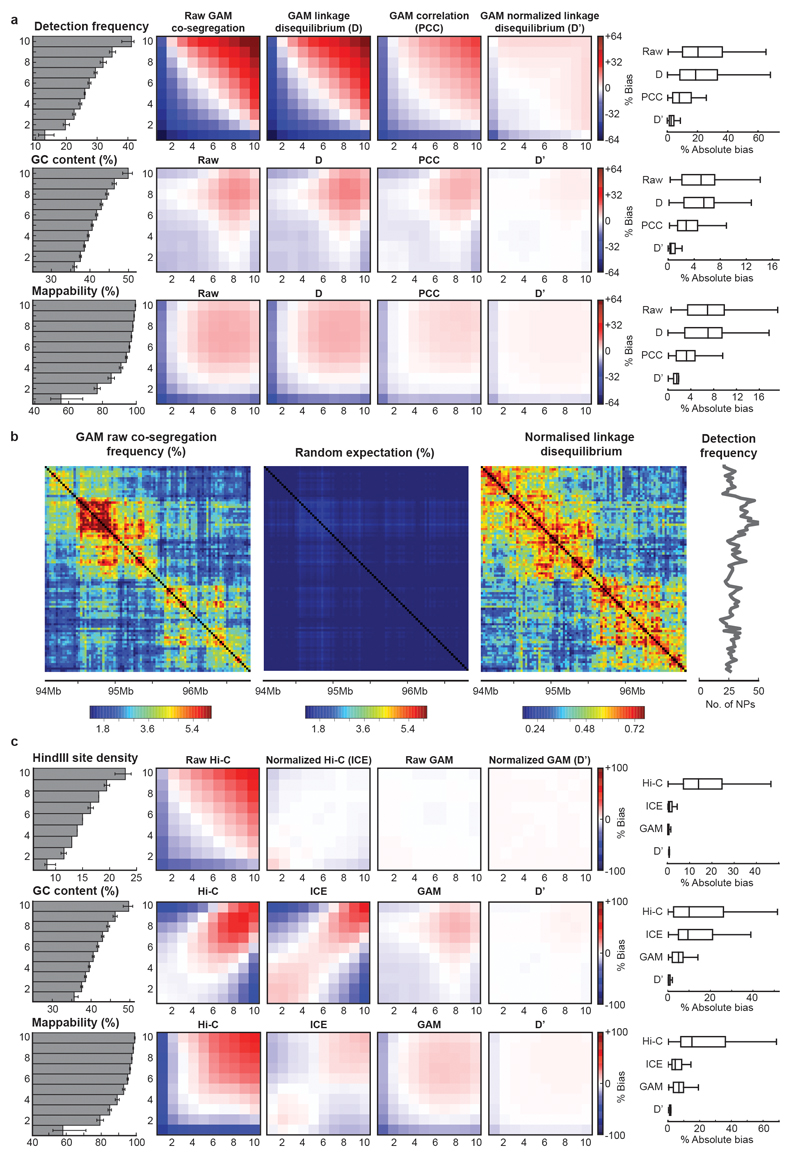
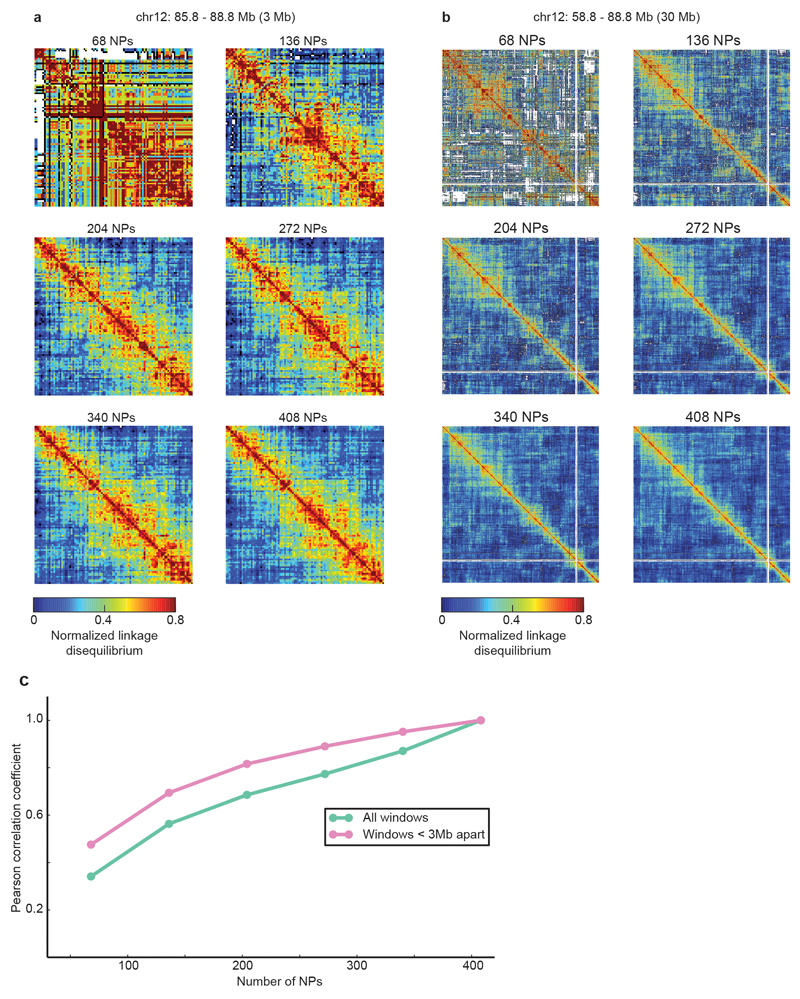
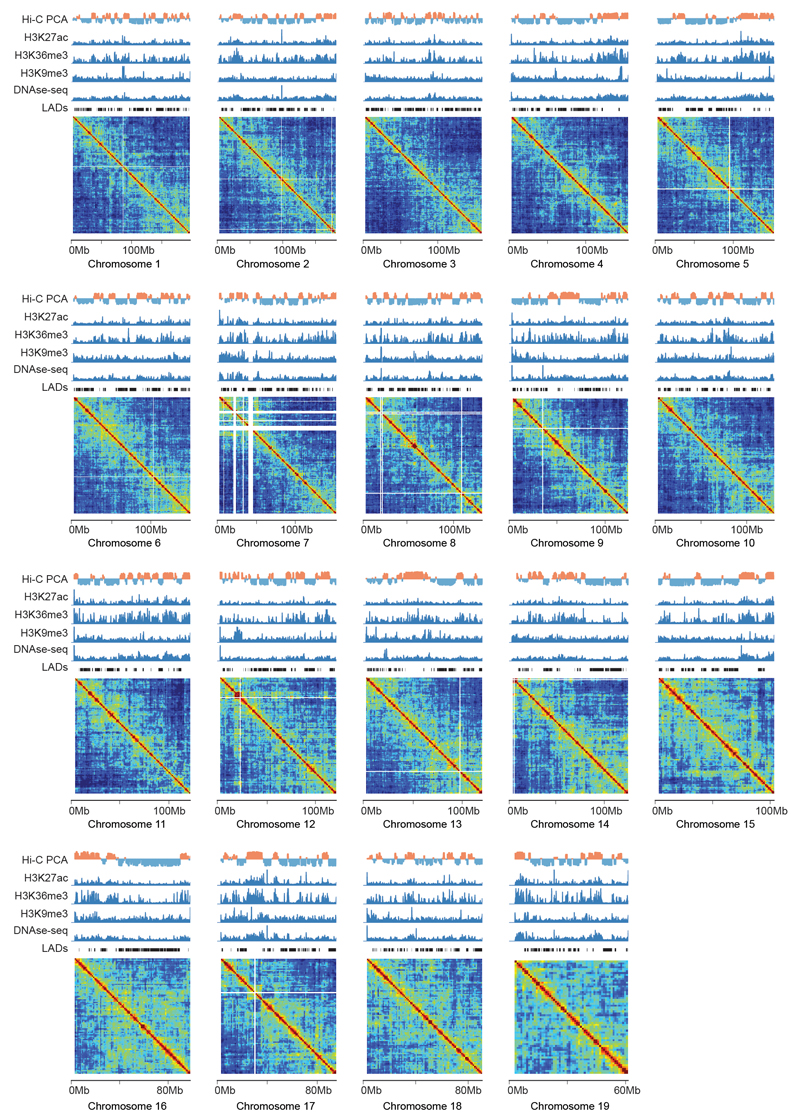
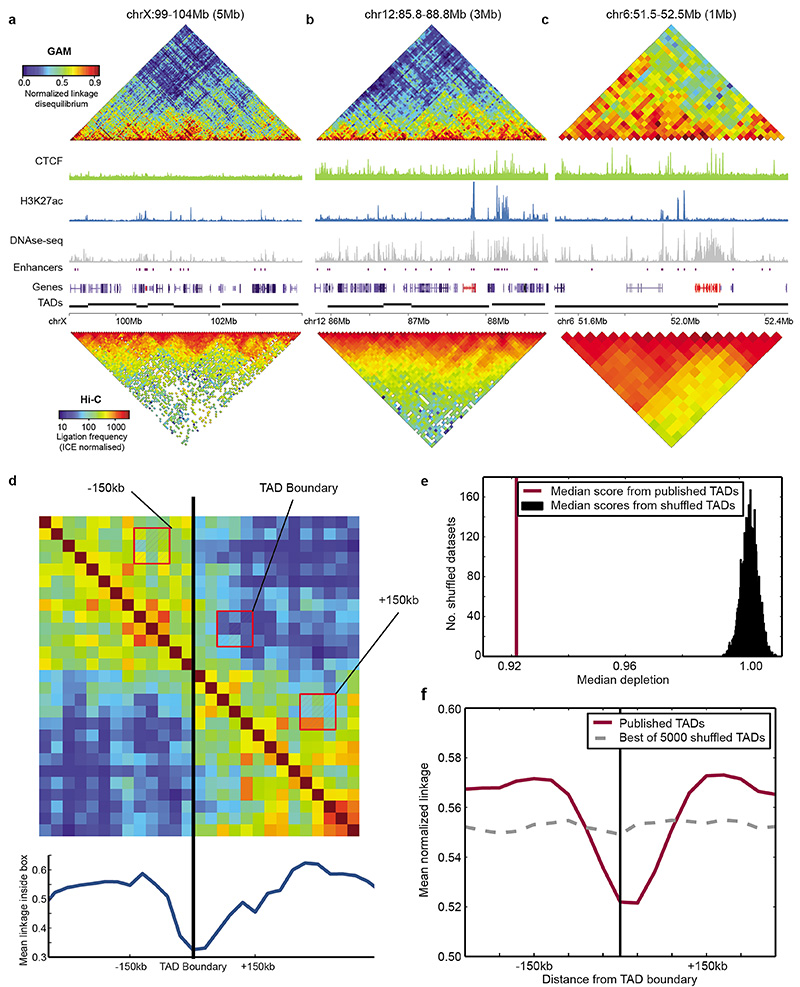
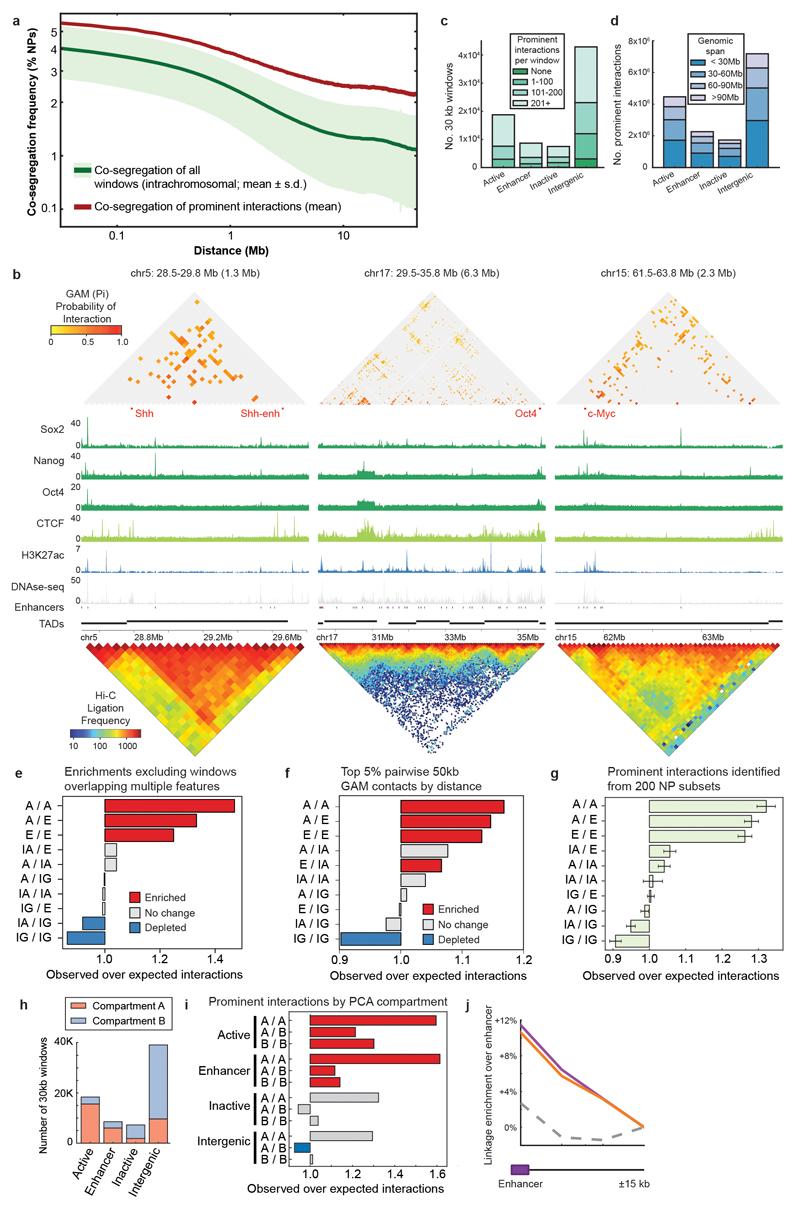
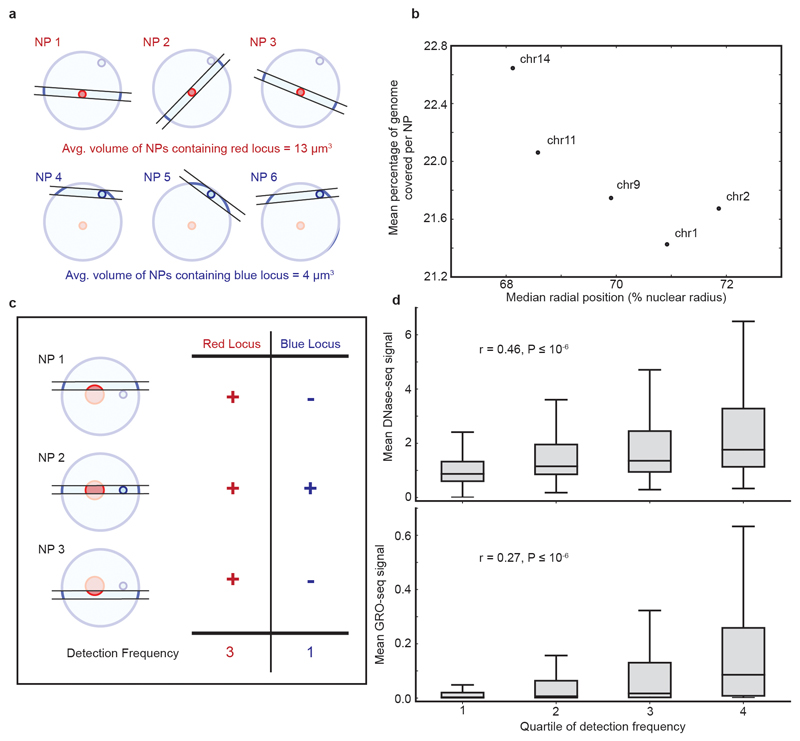
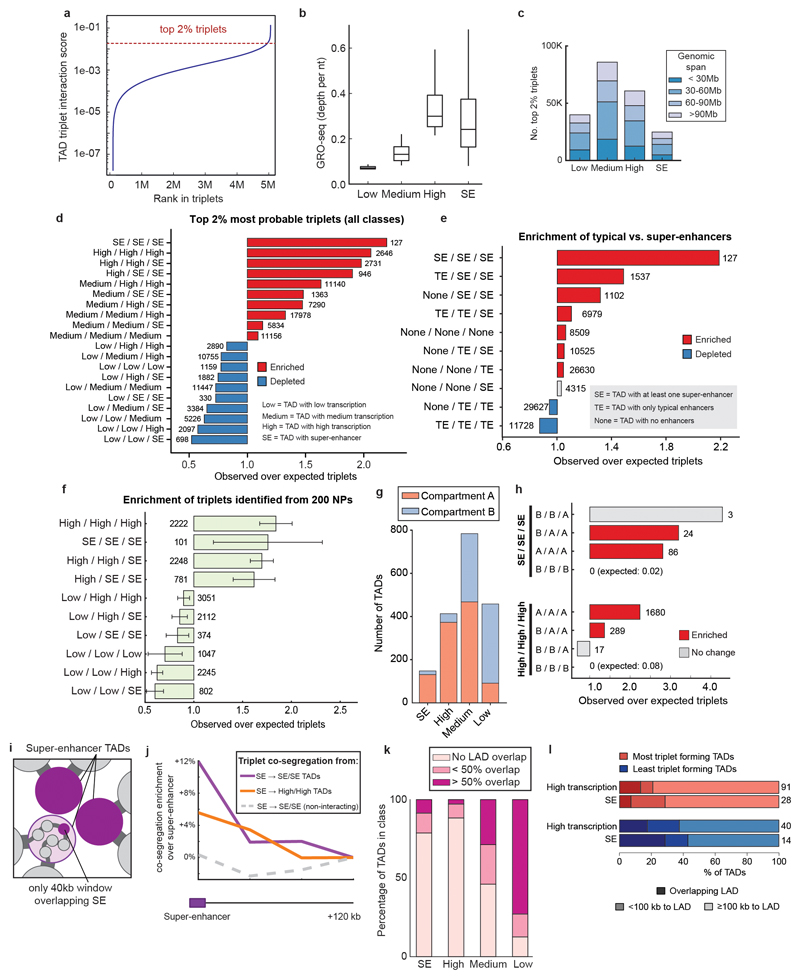
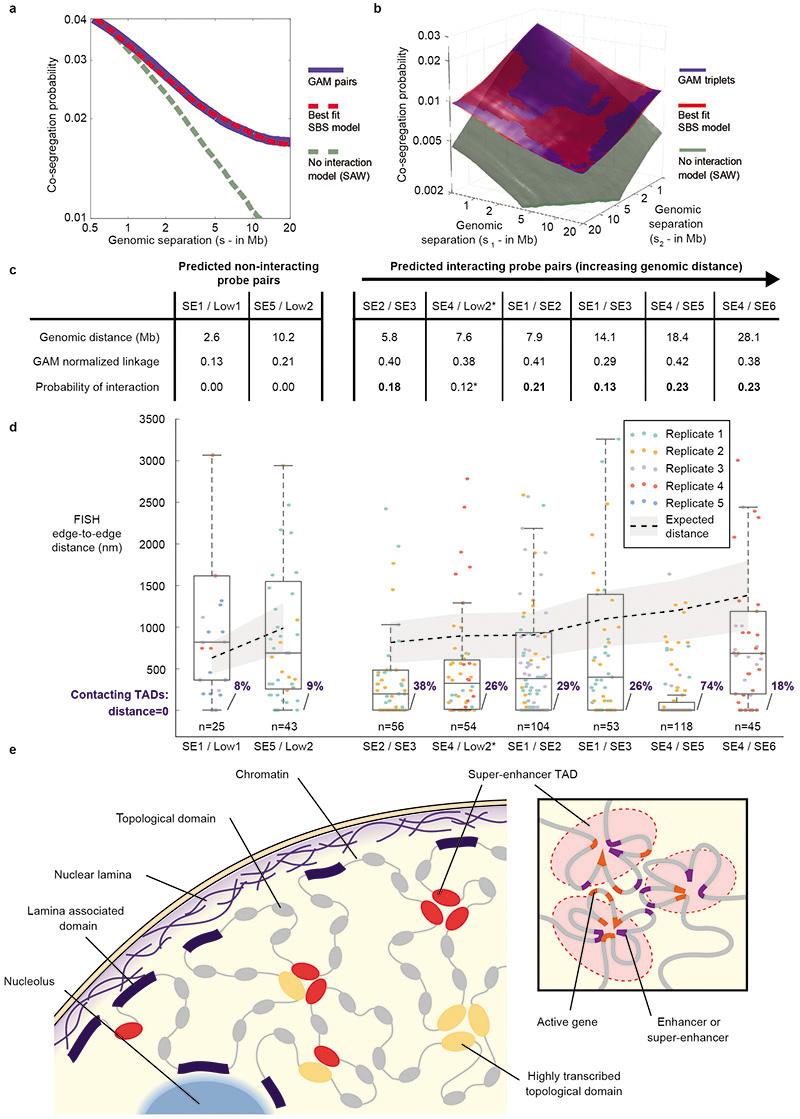
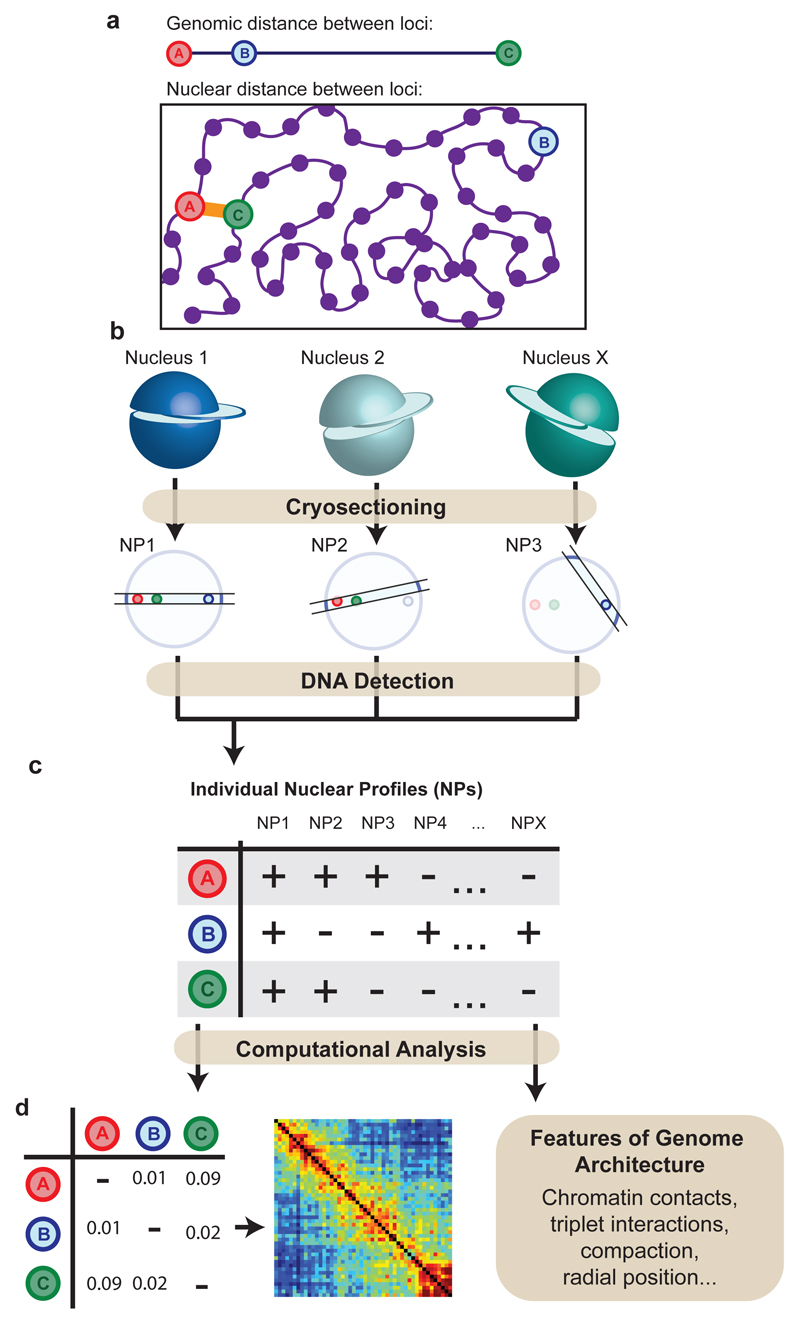
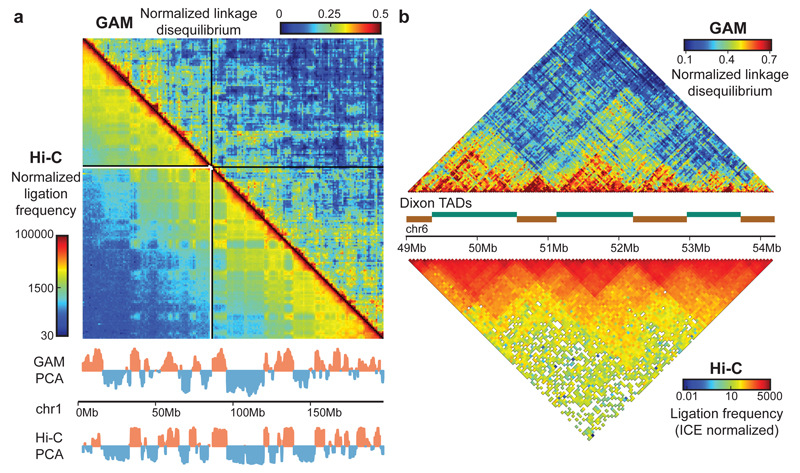
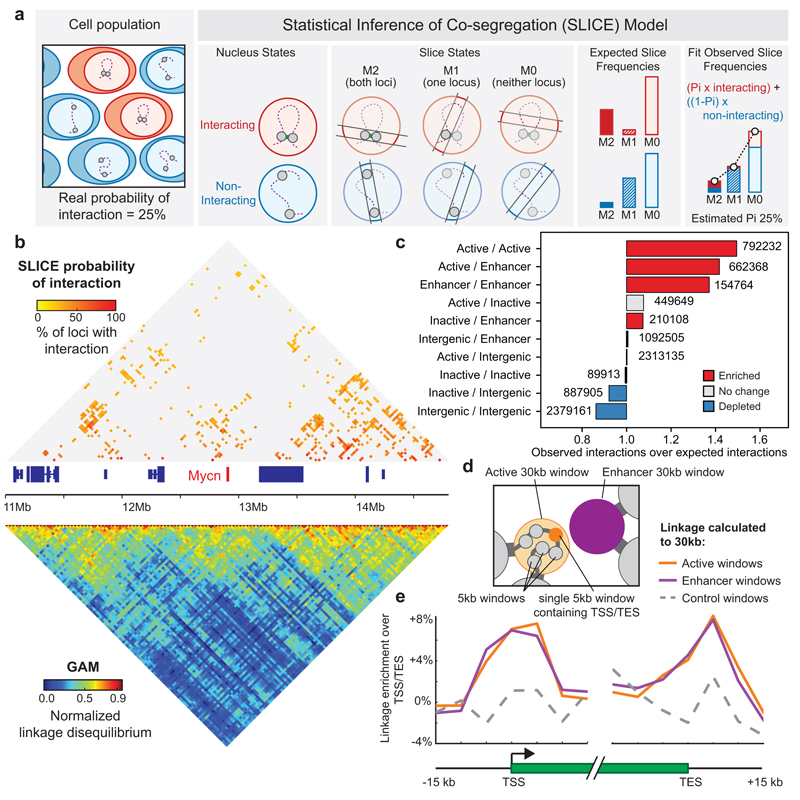
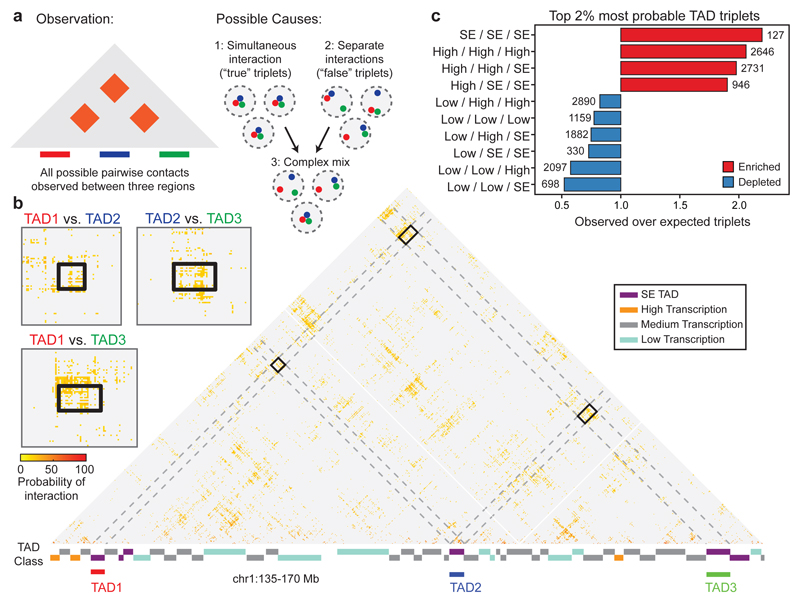
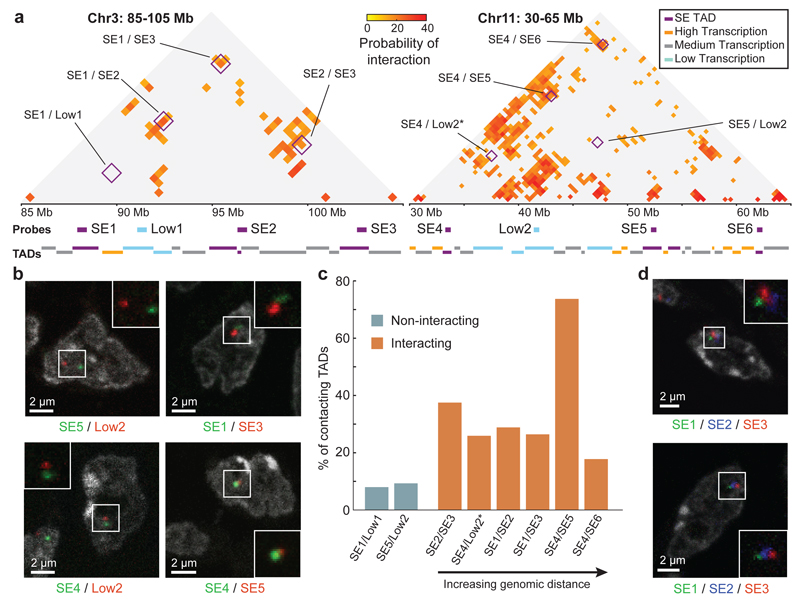
The organization of the genome in the nucleus and the interactions of genes with their regulatory elements are key features of transcriptional control and their disruption can cause disease. Here we report a genome-wide method, genome architecture mapping (GAM), for measuring chromatin contacts and other features of three-dimensional chromatin topology on the basis of sequencing DNA from a large collection of thin nuclear sections. We apply GAM to mouse embryonic stem cells and identify enrichment for specific interactions between active genes and enhancers across very large genomic distances using a mathematical model termed SLICE (statistical inference of co-segregation). GAM also reveals an abundance of three-way contacts across the genome, especially between regions that are highly transcribed or contain super-enhancers, providing a level of insight into genome architecture that, owing to the technical limitations of current technologies, has previously remained unattainable. Furthermore, GAM highlights a role for gene-expression-specific contacts in organizing the genome in mammalian nuclei.
Conflict of interest statement
The authors declare competing financial interests: a patent was filed on behalf of A.P., P.A.W.E., M.N., A.S. and R.A.B. by the Max-Delbrück Centre for Molecular Medicine, Berlin. The other authors declare no competing financial interests.
Figures

Comment in
-
Genome Organization: Zooming in on nuclear organization.Nat Rev Mol Cell Biol. 2017 May;18(5):275. doi: 10.1038/nrm.2017.28. Epub 2017 Mar 22. Nat Rev Mol Cell Biol. 2017. PMID: 28327555 No abstract available.
-
Genome organization: Zooming in on nuclear organization.Nat Rev Genet. 2017 May;18(5):269. doi: 10.1038/nrg.2017.23. Epub 2017 Mar 27. Nat Rev Genet. 2017. PMID: 28344343 No abstract available.
-
Genome Architecture from a Different Angle.Dev Cell. 2017 Apr 10;41(1):3-4. doi: 10.1016/j.devcel.2017.03.017. Dev Cell. 2017. PMID: 28399397 Free PMC article.
-
Genome organization by the slice.Nat Biotechnol. 2017 May 9;35(5):430. doi: 10.1038/nbt.3866. Nat Biotechnol. 2017. PMID: 28486452 No abstract available.
References
-
- Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–11. - PubMed
-
- Simonis M, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–54. - PubMed
-
- Rodley CDM, Bertels F, Jones B, O’Sullivan JM. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genet Biol. 2009;46:879–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
