

Sparse Proteomics Analysis - a compressed sensing-based approach for feature selection and classification of high-dimensional proteomics mass spectrometry data
- PMID: 28274197
- PMCID: PMC5343371
- DOI: 10.1186/s12859-017-1565-4
Sparse Proteomics Analysis - a compressed sensing-based approach for feature selection and classification of high-dimensional proteomics mass spectrometry data
Abstract
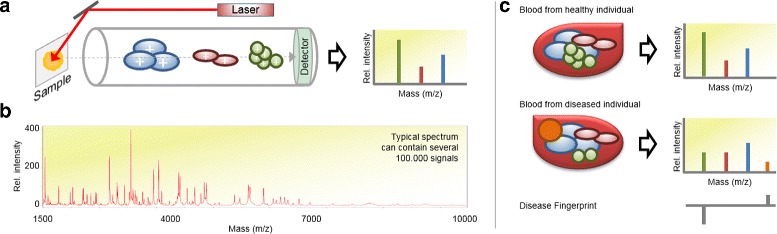
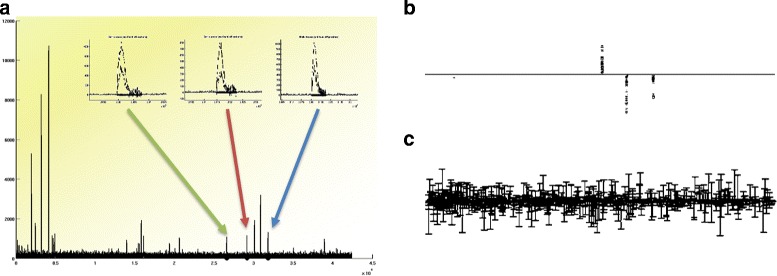
Background: High-throughput proteomics techniques, such as mass spectrometry (MS)-based approaches, produce very high-dimensional data-sets. In a clinical setting one is often interested in how mass spectra differ between patients of different classes, for example spectra from healthy patients vs. spectra from patients having a particular disease. Machine learning algorithms are needed to (a) identify these discriminating features and (b) classify unknown spectra based on this feature set. Since the acquired data is usually noisy, the algorithms should be robust against noise and outliers, while the identified feature set should be as small as possible.
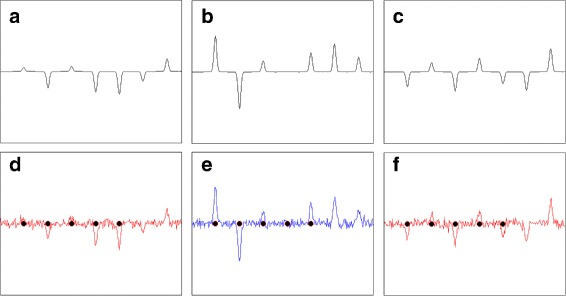
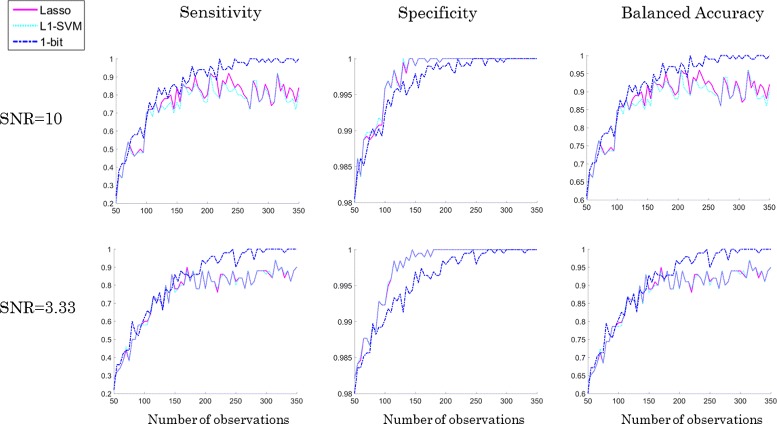
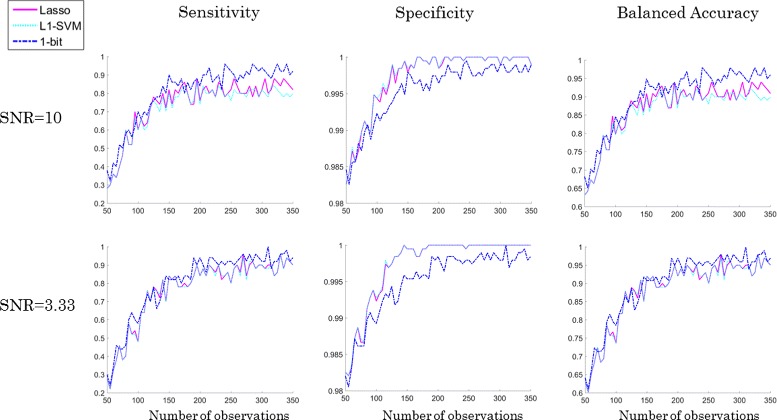
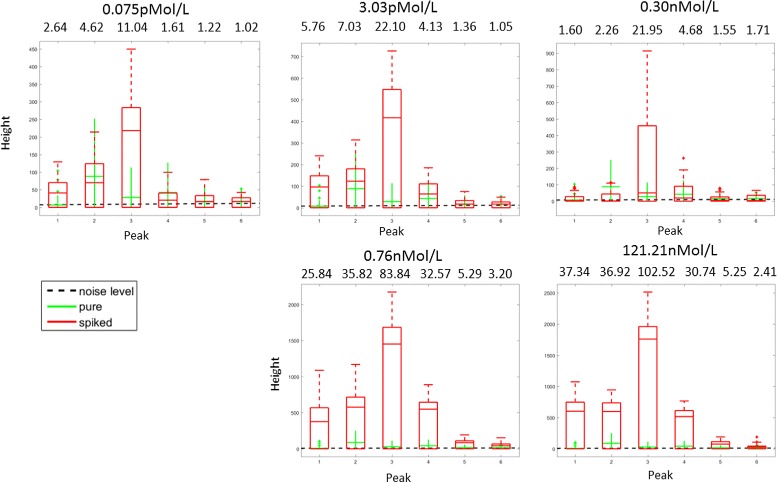
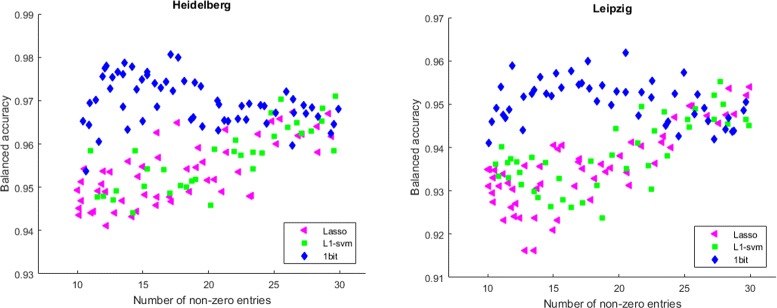
Results: We present a new algorithm, Sparse Proteomics Analysis (SPA), based on the theory of compressed sensing that allows us to identify a minimal discriminating set of features from mass spectrometry data-sets. We show (1) how our method performs on artificial and real-world data-sets, (2) that its performance is competitive with standard (and widely used) algorithms for analyzing proteomics data, and (3) that it is robust against random and systematic noise. We further demonstrate the applicability of our algorithm to two previously published clinical data-sets.
Keywords: Biomarker; Classification; Clinical data; Compressed sensing; Feature selection; Machine learning; Mass spectrometry; Proteomics; Sparsity.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
