

Eteplirsen in the treatment of Duchenne muscular dystrophy
- PMID: 28280301
- PMCID: PMC5338848
- DOI: 10.2147/DDDT.S97635
Eteplirsen in the treatment of Duchenne muscular dystrophy
Abstract
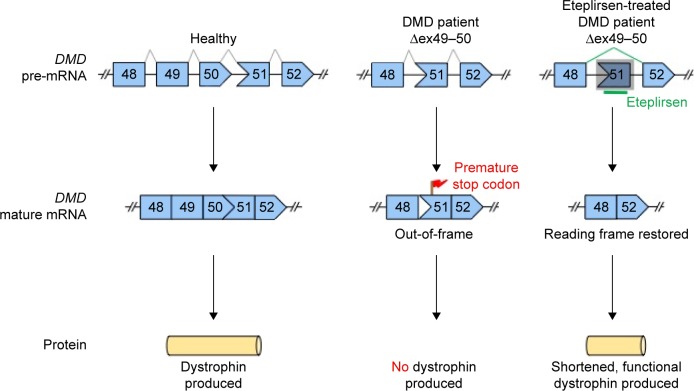
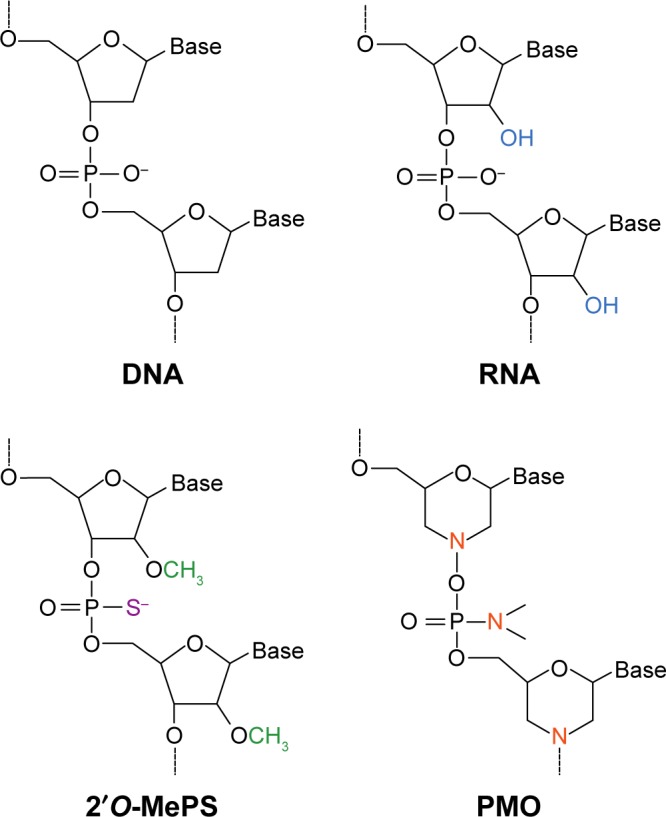
Duchenne muscular dystrophy is a fatal neuromuscular disorder affecting around one in 3,500-5,000 male births that is characterized by progressive muscular deterioration. It is inherited in an X-linked recessive fashion and is caused by loss-of-function mutations in the DMD gene coding for dystrophin, a cytoskeletal protein that stabilizes the plasma membrane of muscle fibers. In September 2016, the US Food and Drug Administration granted accelerated approval for eteplirsen (or Exondys 51), a drug that acts to promote dystrophin production by restoring the translational reading frame of DMD through specific skipping of exon 51 in defective gene variants. Eteplirsen is applicable for approximately 14% of patients with DMD mutations. This article extensively reviews and discusses the available information on eteplirsen to date, focusing on pharmacological, efficacy, safety, and tolerability data from preclinical and clinical trials. Issues faced by eteplirsen, particularly those relating to its efficacy, will be identified. Finally, the place of eteplirsen and exon skipping as a general therapeutic strategy in Duchenne muscular dystrophy treatment will be discussed.
Keywords: Duchenne muscular dystrophy; Exondys 51; eteplirsen; exon-skipping therapy; phosphorodiamidate morpholino oligomers.
Conflict of interest statement
Disclosure The authors report no conflicts of interest in this work.
Figures
References
-
- Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. - PubMed
-
- Emery AE. Population frequencies of inherited neuromuscular diseases: a world survey. Neuromuscul Disord. 1991;1(1):19–29. - PubMed
-
- Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–313. - PubMed
-
- Manzur A, Kinali M, Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child. 2008;93(11):986–990. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
