

Existence of HbF Enhancer Haplotypes at HBS1L-MYB Intergenic Region in Transfusion-Dependent Saudi β-Thalassemia Patients
- PMID: 28280727
- PMCID: PMC5322420
- DOI: 10.1155/2017/1972429
Existence of HbF Enhancer Haplotypes at HBS1L-MYB Intergenic Region in Transfusion-Dependent Saudi β-Thalassemia Patients
Abstract
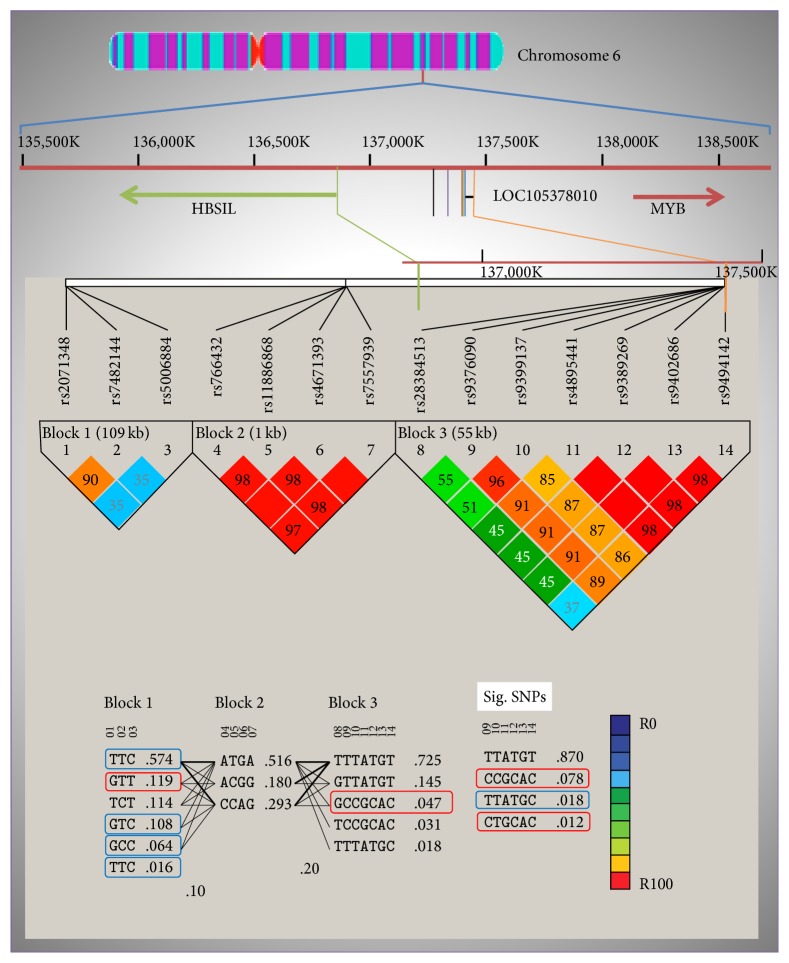
Background and Objectives. β-Thalassemia and sickle cell disease are genetic disorders characterized by reduced and abnormal β-globin chain production, respectively. The elevation of fetal hemoglobin (HbF) can ameliorate the severity of these disorders. In sickle cell disease patients, the HbF level elevation is associated with three quantitative trait loci (QTLs), BCL11A, HBG2 promoter, and HBS1L-MYB intergenic region. This study elucidates the existence of the variants in these three QTLs to determine their association with HbF levels of transfusion-dependent Saudi β-thalassemia patients. Materials and Methods. A total of 174 transfusion-dependent β-thalassemia patients and 164 healthy controls from Eastern Province of Saudi Arabia were genotyped for fourteen single nucleotide polymorphisms (SNPs) from the three QTL regions using TaqMan assay on real-time PCR. Results. Genotype analysis revealed that six alleles of HBS1L-MYB QTL (rs9376090C p = 0.0009, rs9399137C p = 0.008, rs4895441G p = 0.004, rs9389269C p = 0.008, rs9402686A p = 0.008, and rs9494142C p = 0.002) were predominantly associated with β-thalassemia. In addition, haplotype analysis revealed that haplotypes of HBS1L-MYB (GCCGCAC p = 0.022) and HBG2 (GTT p = 0.009) were also predominantly associated with β-thalassemia. Furthermore, the HBS1L-MYB region also exhibited association with the high HbF cohort. Conclusion. The stimulation of HbF gene expression may provide alternative therapies for the amelioration of the disease severity of β-thalassemia.
Conflict of interest statement
The authors report no conflict of interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
