

Proteasome inhibitors against amelanotic melanoma
- PMID: 28281027
- PMCID: PMC5658467
- DOI: 10.1007/s10565-017-9390-0
Proteasome inhibitors against amelanotic melanoma
Abstract
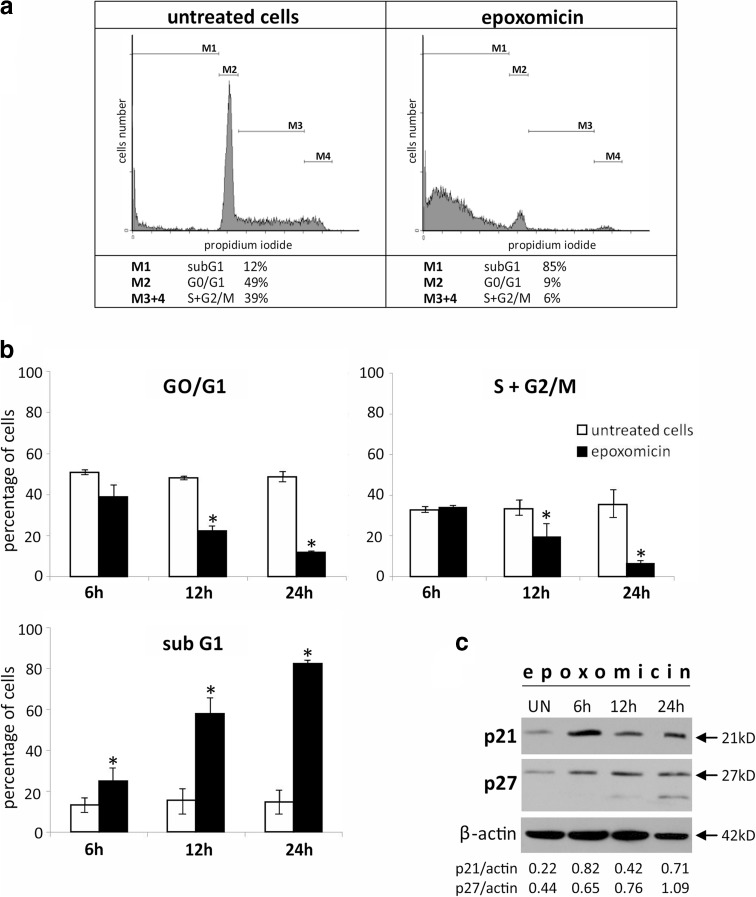
The incidence of malignant melanoma, the most aggressive skin cancer, is increasing constantly. Despite new targeted therapies, the prognosis for patients with metastatic disease remains poor. Thus, there is a need for new combinational treatments, and antineoplastic agents potentially valuable in this approach are inhibitors of the ubiquitin-proteasome system (UPS). In this work, we analyze the cytotoxicity mechanisms of proteasome inhibitors (MG-132, epoxomicin, and lactacystin) in a specific form of melanoma which does not synthesize melanin-the amelanotic melanoma (Ab cells). We found that the most cytotoxic of the compounds tested was epoxomicin. Caspase-9 activation as well as cytochrome C and AIF release from mitochondria indicated that exposure to epoxomicin induced the mitochondrial pathway of apoptosis. Epoxomicin treatment also resulted in accumulation of Bcl-2 family members-proapoptotic Noxa and antiapoptotic Mcl-1, which were postulated as the targets for bortezomib in melanoma. Inhibition of caspases by BAF revealed that cell death was partially caspase-independent. We observed no cell cycle arrest preceding the apoptosis of Ab cells, even though cdk inhibitors p21Cip1/Waf1 and p27Kip1 were up-regulated. The cell cycle was blocked only after inactivation of caspases by the pan-caspase inhibitor BAF. In summary, this is the first study exploring molecular mechanisms of cell death induced by epoxomicin in melanoma. We found that Ab cells died on the mitochondrial pathway of apoptosis and also partially by the caspase-independent way of death. Apoptosis induction was fast and efficient and was not preceded by cell cycle arrest.
Keywords: Apoptosis; Cancer; Melanoma; Proteasome inhibitors.
Figures





Similar articles
-
The activity of caspases in spontaneous and camptothecin-induced death of melanotic and amelanotic melanoma cell.Cancer Biol Ther. 2007 Mar;6(3):346-53. doi: 10.4161/cbt.6.3.3701. Epub 2007 Mar 12. Cancer Biol Ther. 2007. PMID: 17312383
-
Camptothecin-induced death of amelanotic and melanotic melanoma cells in different phases of cell cycle.Neoplasma. 2011;58(3):227-34. doi: 10.4149/neo_2011_03_227. Neoplasma. 2011. PMID: 21391739
-
Cooperative cytotoxicity of proteasome inhibitors and tumor necrosis factor-related apoptosis-inducing ligand in chemoresistant Bcl-2-overexpressing cells.Clin Cancer Res. 2005 Jun 1;11(11):4259-65. doi: 10.1158/1078-0432.CCR-04-2496. Clin Cancer Res. 2005. PMID: 15930365
-
Mechanisms of apoptosis induced by anticancer compounds in melanoma cells.Curr Top Med Chem. 2012;12(1):50-2. doi: 10.2174/156802612798919196. Curr Top Med Chem. 2012. PMID: 22280162 Review.
-
Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance.Drug Resist Updat. 2020 Jan;48:100663. doi: 10.1016/j.drup.2019.100663. Epub 2019 Nov 11. Drug Resist Updat. 2020. PMID: 31785545 Review.
Cited by
-
Amelanotic Melanoma-Biochemical and Molecular Induction Pathways.Int J Mol Sci. 2024 Oct 26;25(21):11502. doi: 10.3390/ijms252111502. Int J Mol Sci. 2024. PMID: 39519055 Free PMC article. Review.
-
The role of the ubiquitin-proteasome pathway in skin cancer development: 26S proteasome-activated NF-κB signal transduction.Cancer Biol Ther. 2021 Dec 2;22(10-12):479-492. doi: 10.1080/15384047.2021.1978785. Epub 2021 Sep 29. Cancer Biol Ther. 2021. PMID: 34583610 Free PMC article.
-
The interconnective role of the UPS and autophagy in the quality control of cancer mitochondria.Cell Mol Life Sci. 2025 Jan 12;82(1):42. doi: 10.1007/s00018-024-05556-x. Cell Mol Life Sci. 2025. PMID: 39800773 Free PMC article. Review.
-
KIFC3 Promotes Proliferation, Migration, and Invasion in Colorectal Cancer via PI3K/AKT/mTOR Signaling Pathway.Front Genet. 2022 Jun 22;13:848926. doi: 10.3389/fgene.2022.848926. eCollection 2022. Front Genet. 2022. PMID: 35812733 Free PMC article.
-
Equine Melanocytic Tumors: A Narrative Review.Animals (Basel). 2023 Jan 10;13(2):247. doi: 10.3390/ani13020247. Animals (Basel). 2023. PMID: 36670786 Free PMC article. Review.
References
-
- Amann VC, Ramelyte E, Thurneysen S, Pitocco R, Bentele-Jaberg N, Goldinger SM, et al. Developments in targeted therapy in melanoma. Eur J Surg Oncol. 2016:1–13. - PubMed
-
- Amschler K, Schön MP, Pletz N, Wallbrecht K, Erpenbeck L, Schön M. NF-kappa B inhibition through proteasome inhibition or IKKbeta blockade increases the susceptibility of melanoma cells to cytostatic treatment through distinct pathways. J Invest Dermatol. 2010;130:1073–1086. doi: 10.1038/jid.2009.365. - DOI - PubMed
-
- Berenson A, Vardanyan S, David M, Wang J, Harutyunyan NM, Gottlieb J, et al. Outcomes of multiple myeloma patients receiving bortezomib, lenalidomide, and carfilzomib. Ann Hematol. 2016 (ahead of print) - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
