

Sweet and sour: an update on classic galactosemia
- PMID: 28281081
- PMCID: PMC5391384
- DOI: 10.1007/s10545-017-0029-3
Sweet and sour: an update on classic galactosemia
Abstract
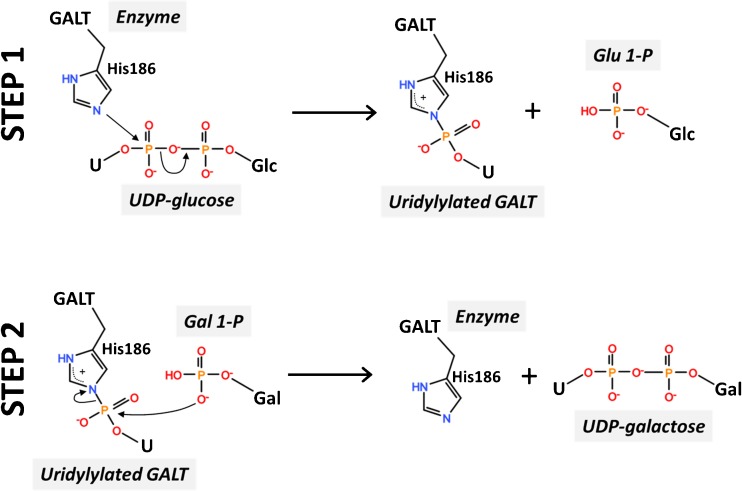
Classic galactosemia is a rare inherited disorder of galactose metabolism caused by deficient activity of galactose-1-phosphate uridylyltransferase (GALT), the second enzyme of the Leloir pathway. It presents in the newborn period as a life-threatening disease, whose clinical picture can be resolved by a galactose-restricted diet. The dietary treatment proves, however, insufficient in preventing severe long-term complications, such as cognitive, social and reproductive impairments. Classic galactosemia represents a heavy burden on patients' and their families' lives. After its first description in 1908 and despite intense research in the past century, the exact pathogenic mechanisms underlying galactosemia are still not fully understood. Recently, new important insights on molecular and cellular aspects of galactosemia have been gained, and should open new avenues for the development of novel therapeutic strategies. Moreover, an international galactosemia network has been established, which shall act as a platform for expertise and research in galactosemia. Herein are reviewed some of the latest developments in clinical practice and research findings on classic galactosemia, an enigmatic disorder with many unanswered questions warranting dedicated research.
Conflict of interest statement
Conflict of interest
None.
Informed consent/animal rights
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Funding
iNOVA4Health - UID/Multi/04462/2013, a program financially supported by FCT/Ministério da Educação e Ciência, through national funds and co-funded by FEDER under the PT2020 Partnership Agreement, is acknowledged. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
