

Bayesian molecular design with a chemical language model
- PMID: 28281211
- PMCID: PMC5393296
- DOI: 10.1007/s10822-016-0008-z
Bayesian molecular design with a chemical language model
Abstract
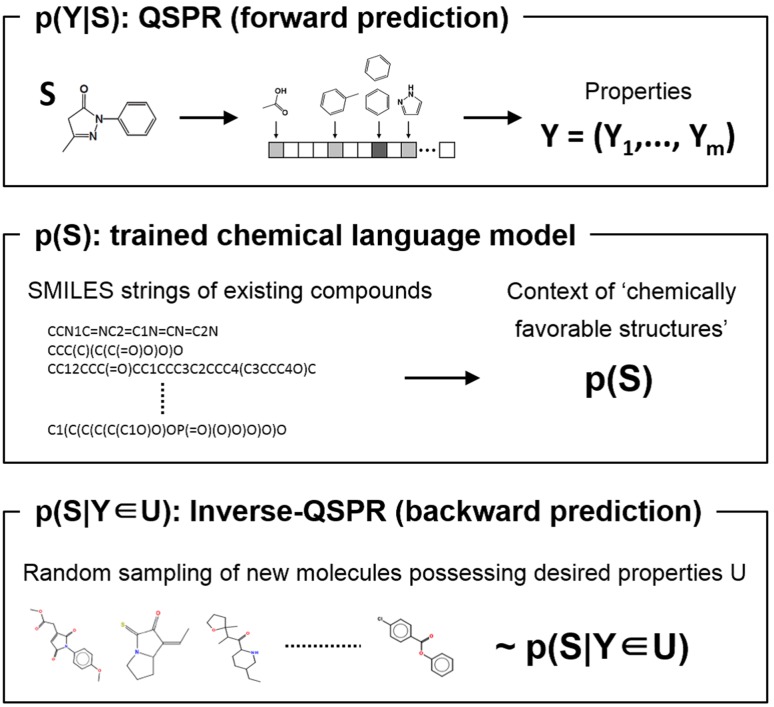
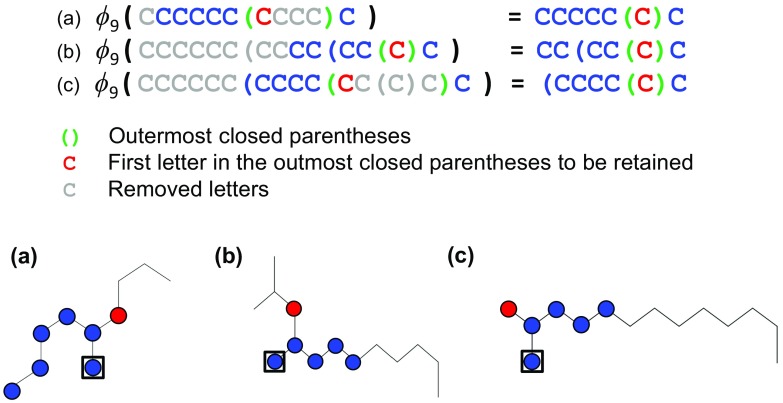
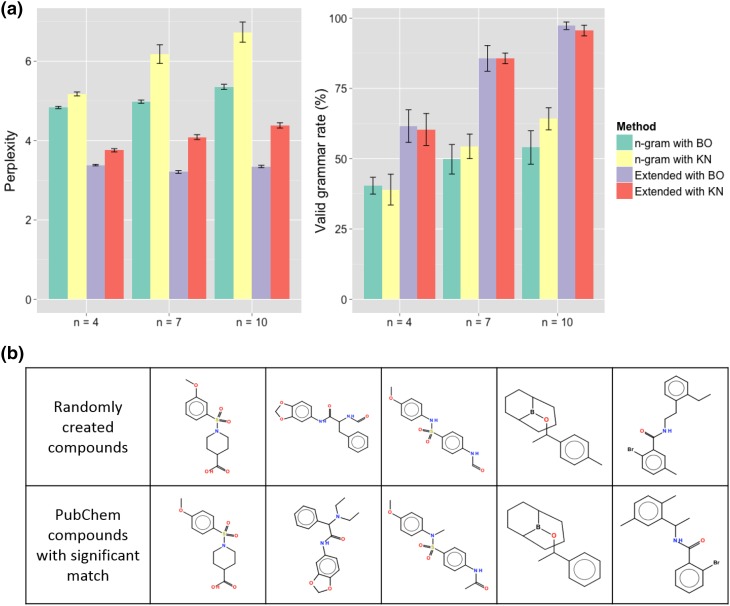
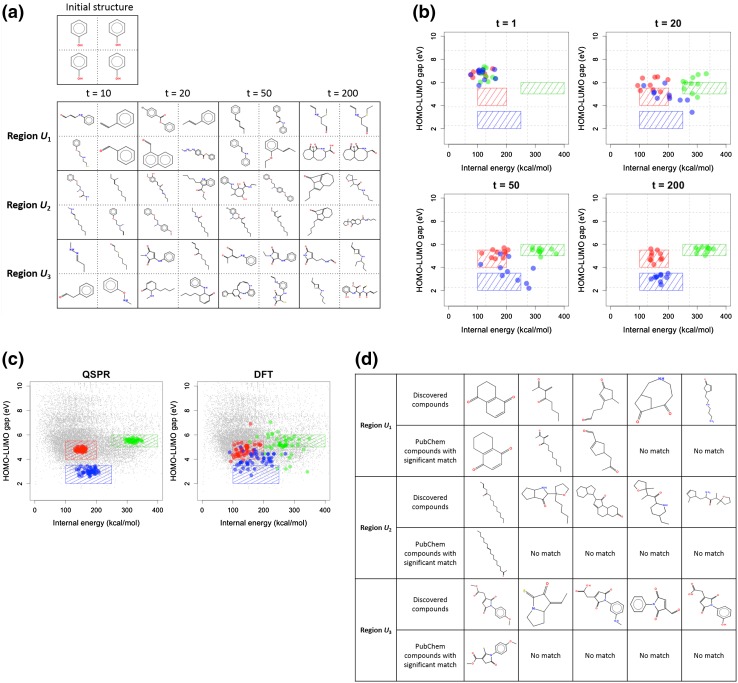
The aim of computational molecular design is the identification of promising hypothetical molecules with a predefined set of desired properties. We address the issue of accelerating the material discovery with state-of-the-art machine learning techniques. The method involves two different types of prediction; the forward and backward predictions. The objective of the forward prediction is to create a set of machine learning models on various properties of a given molecule. Inverting the trained forward models through Bayes' law, we derive a posterior distribution for the backward prediction, which is conditioned by a desired property requirement. Exploring high-probability regions of the posterior with a sequential Monte Carlo technique, molecules that exhibit the desired properties can computationally be created. One major difficulty in the computational creation of molecules is the exclusion of the occurrence of chemically unfavorable structures. To circumvent this issue, we derive a chemical language model that acquires commonly occurring patterns of chemical fragments through natural language processing of ASCII strings of existing compounds, which follow the SMILES chemical language notation. In the backward prediction, the trained language model is used to refine chemical strings such that the properties of the resulting structures fall within the desired property region while chemically unfavorable structures are successfully removed. The present method is demonstrated through the design of small organic molecules with the property requirements on HOMO-LUMO gap and internal energy. The R package iqspr is available at the CRAN repository.
Keywords: Bayesian analysis; Inverse-QSPR; Molecular design; Natural language processing; SMILES; Small organic molecules.
Figures
References
-
- Kawashita N, et al. A mini-review on chemoinformatics approaches for drug discovery. J Comput Aided Chem. 2015;16:15–29. doi: 10.2751/jcac.16.15. - DOI
-
- Venkatasubramanian V, Chan K, Caruthers JM. Computer-aided molecular design using genetic algorithms. Comput Chem Eng. 1994;18:833–844. doi: 10.1016/0098-1354(93)E0023-3. - DOI
-
- Venkatasubramanian V, Chan K, Caruthers JM. Evolutionary design of molecules with desired properties using the genetic algorithm. J Chem Inf Comput Sci. 1995;35:188–195. doi: 10.1021/ci00024a003. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
