

Metafounders are related to F st fixation indices and reduce bias in single-step genomic evaluations
- PMID: 28283016
- PMCID: PMC5439149
- DOI: 10.1186/s12711-017-0309-2
Metafounders are related to F st fixation indices and reduce bias in single-step genomic evaluations
Abstract
Background: Metafounders are pseudo-individuals that encapsulate genetic heterozygosity and relationships within and across base pedigree populations, i.e. ancestral populations. This work addresses the estimation and usefulness of metafounder relationships in single-step genomic best linear unbiased prediction (ssGBLUP).
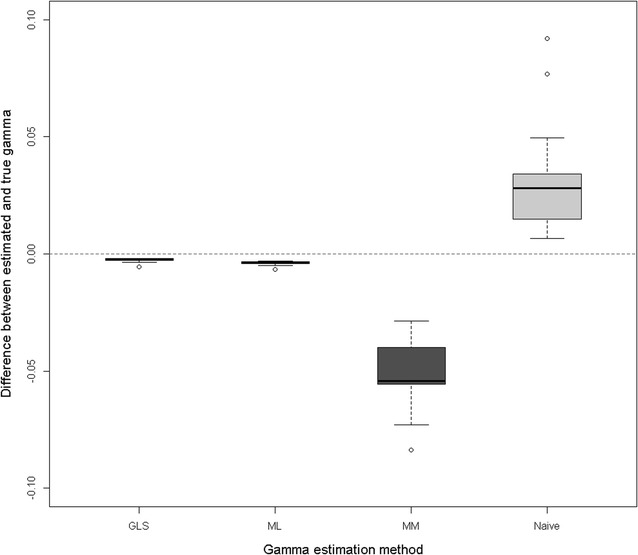
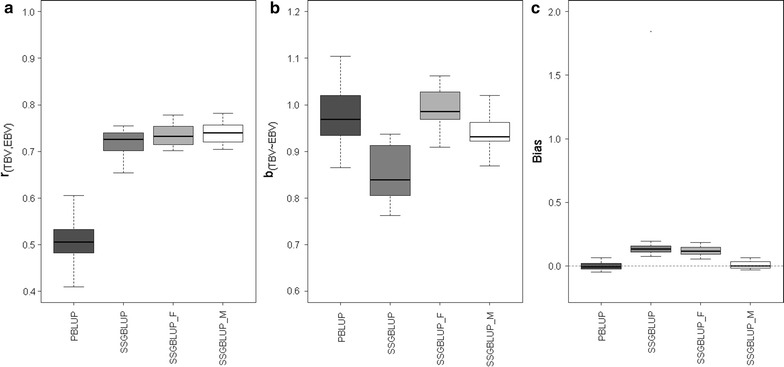
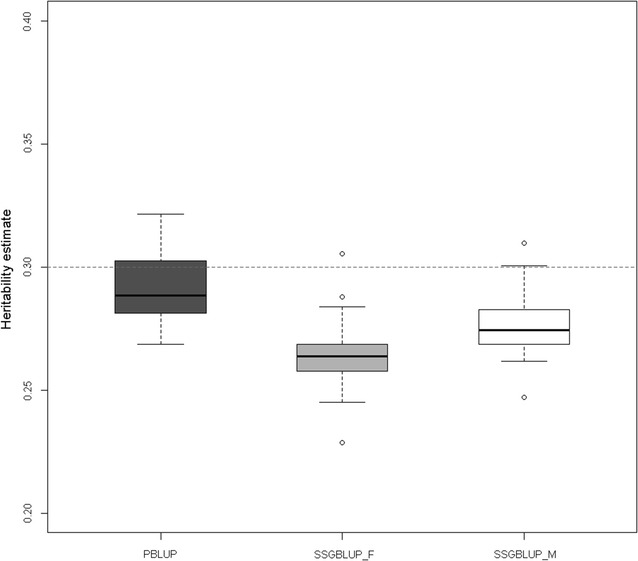
Results: We show that ancestral relationship parameters are proportional to standardized covariances of base allelic frequencies across populations, such as [Formula: see text] fixation indexes. These covariances of base allelic frequencies can be estimated from marker genotypes of related recent individuals and pedigree. Simple methods for their estimation include naïve computation of allele frequencies from marker genotypes or a method of moments that equates average pedigree-based and marker-based relationships. Complex methods include generalized least squares (best linear unbiased estimator (BLUE)) or maximum likelihood based on pedigree relationships. To our knowledge, methods to infer [Formula: see text] coefficients from marker data have not been developed for related individuals. We derived a genomic relationship matrix, compatible with pedigree relationships, that is constructed as a cross-product of {-1,0,1} codes and that is equivalent (apart from scale factors) to an identity-by-state relationship matrix at genome-wide markers. Using a simulation with a single population under selection in which only males and youngest animals are genotyped, we observed that generalized least squares or maximum likelihood gave accurate and unbiased estimates of the ancestral relationship parameter (true value: 0.40) whereas the naïve method and the method of moments were biased (average estimates of 0.43 and 0.35). We also observed that genomic evaluation by ssGBLUP using metafounders was less biased in terms of estimates of genetic trend (bias of 0.01 instead of 0.12), resulted in less overdispersed (0.94 instead of 0.99) and as accurate (0.74) estimates of breeding values than ssGBLUP without metafounders and provided consistent estimates of heritability.
Conclusions: Estimation of metafounder relationships can be achieved using BLUP-like methods with pedigree and markers. Inclusion of metafounder relationships reduces bias of genomic predictions with no loss in accuracy.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
