

Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome
- PMID: 28283040
- PMCID: PMC5346261
- DOI: 10.1186/s13059-017-1173-7
Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome
Erratum in
-
Correction to: Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome.Genome Biol. 2019 May 7;20(1):89. doi: 10.1186/s13059-019-1702-7. Genome Biol. 2019. PMID: 31064398 Free PMC article.
Abstract
Background: The functional impact of genetic variation has been extensively surveyed, revealing that genetic changes correlated to phenotypes lie mostly in non-coding genomic regions. Studies have linked allele-specific genetic changes to gene expression, DNA methylation, and histone marks but these investigations have only been carried out in a limited set of samples.
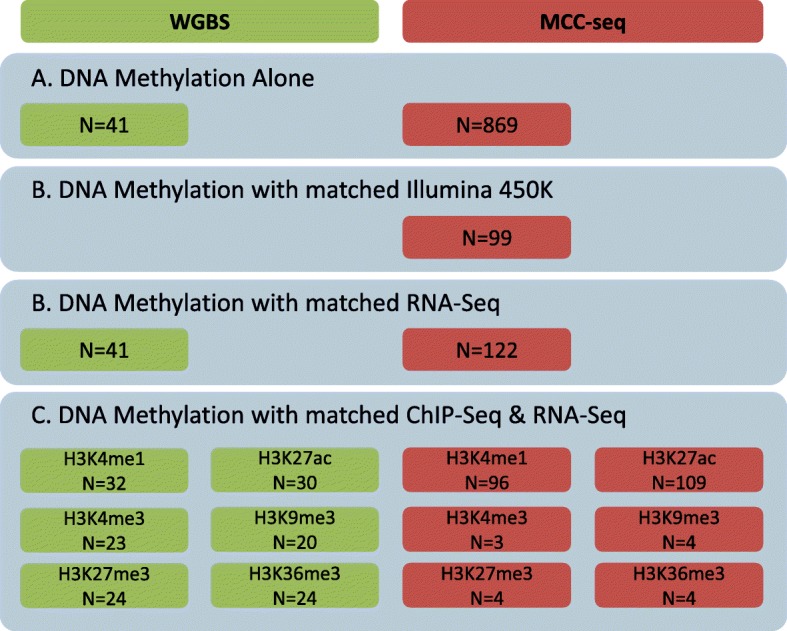
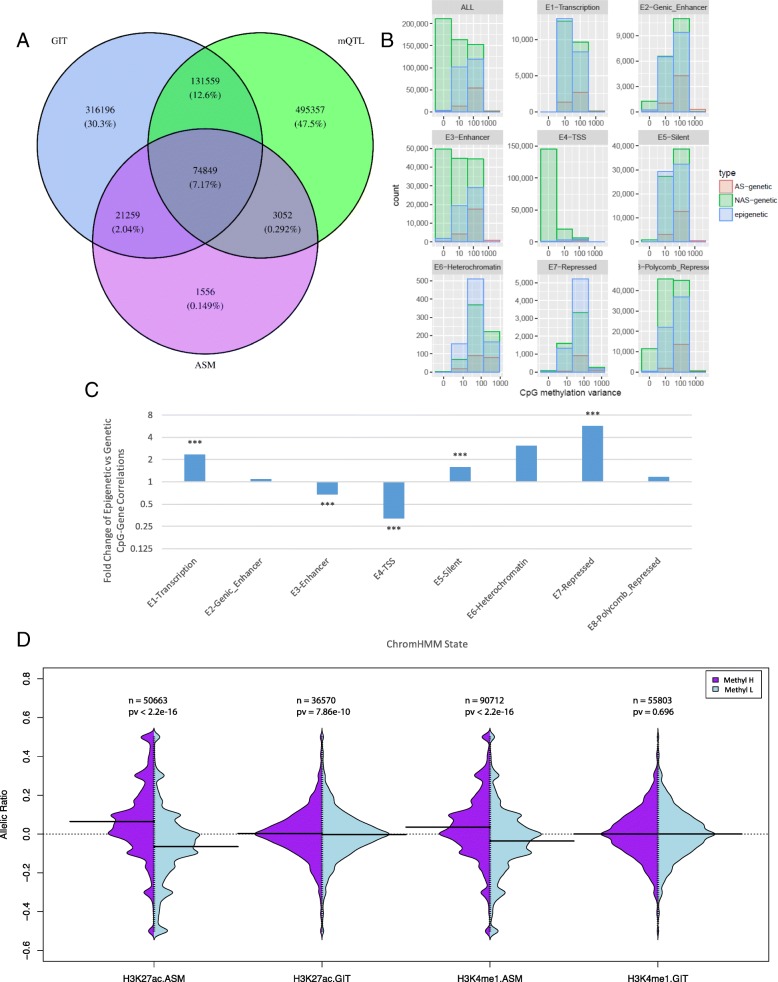
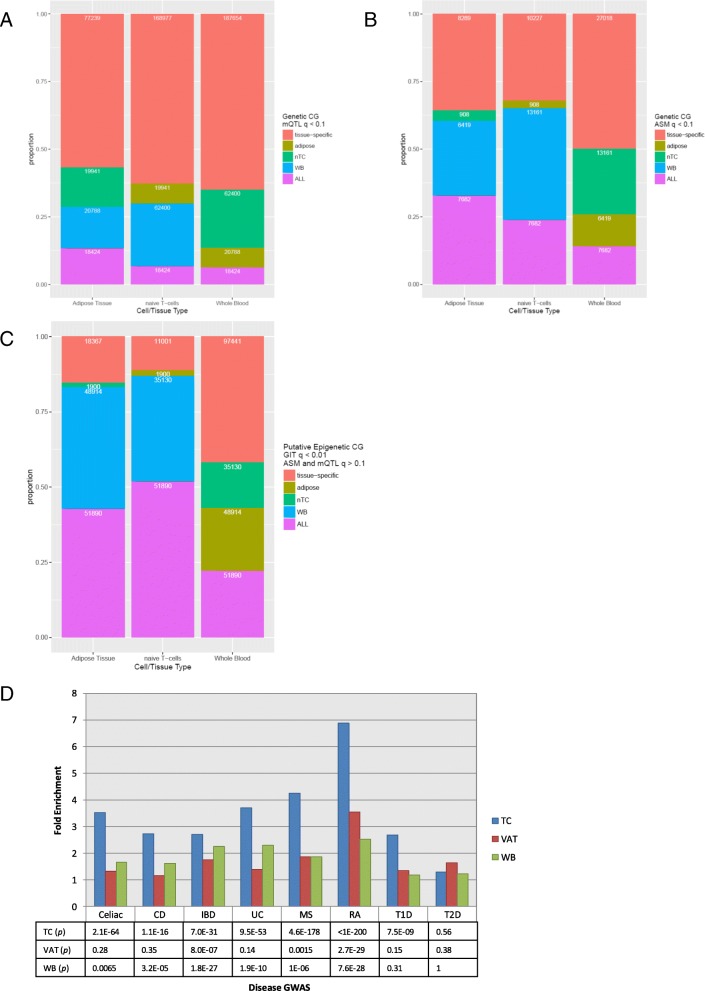
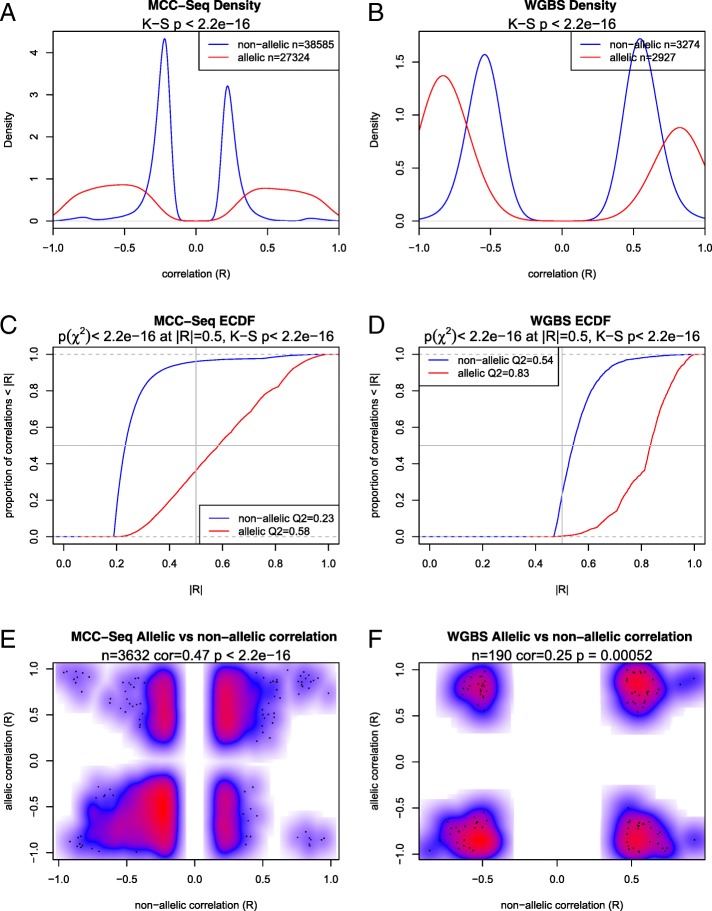
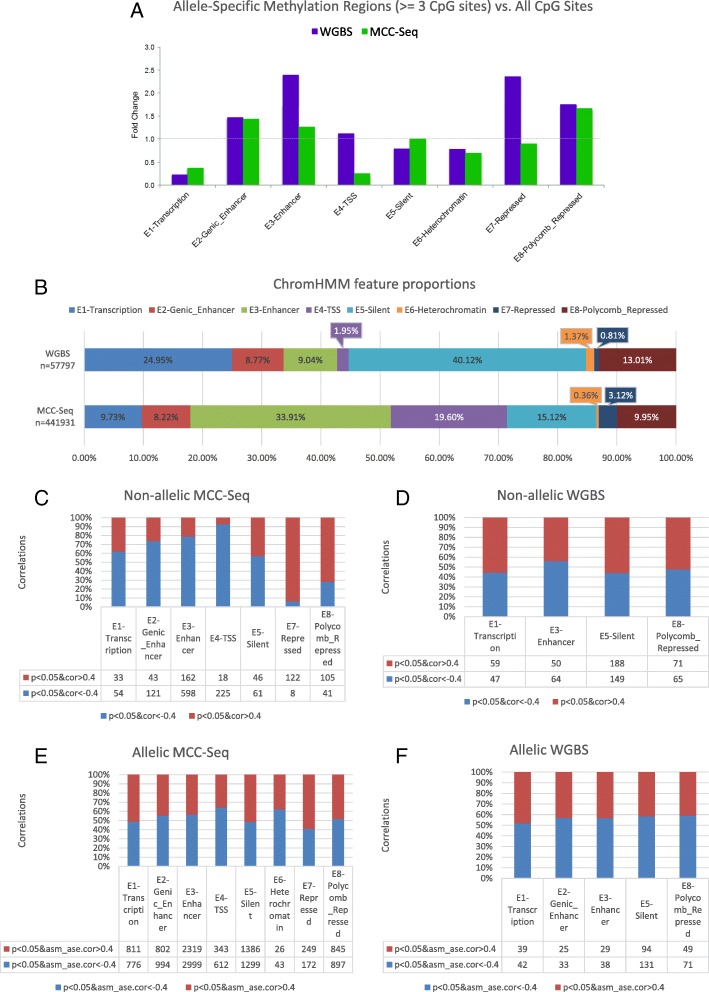
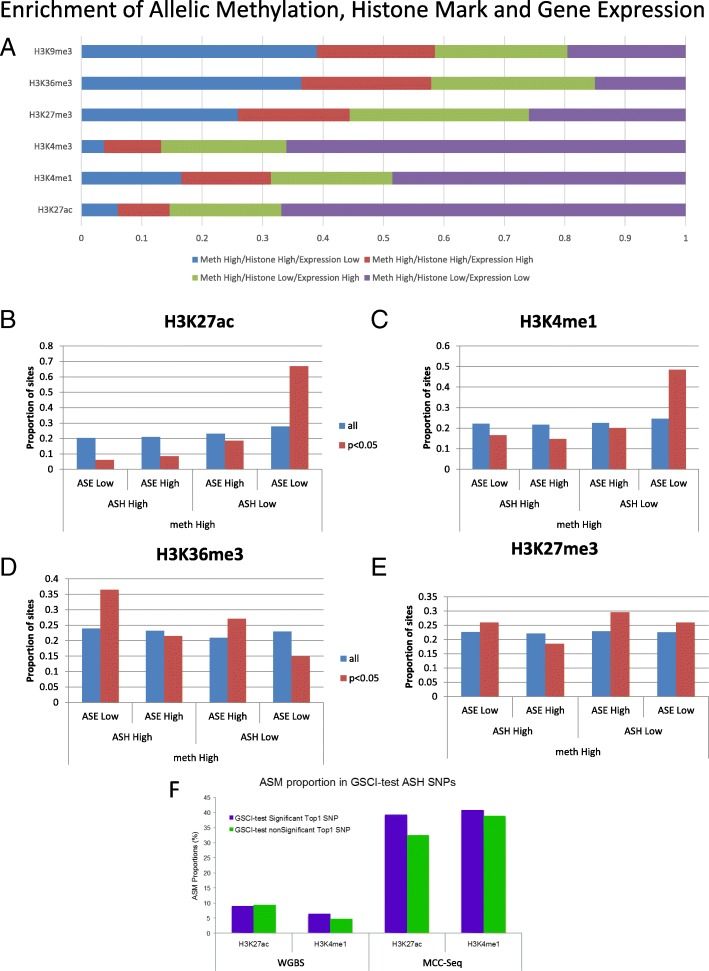
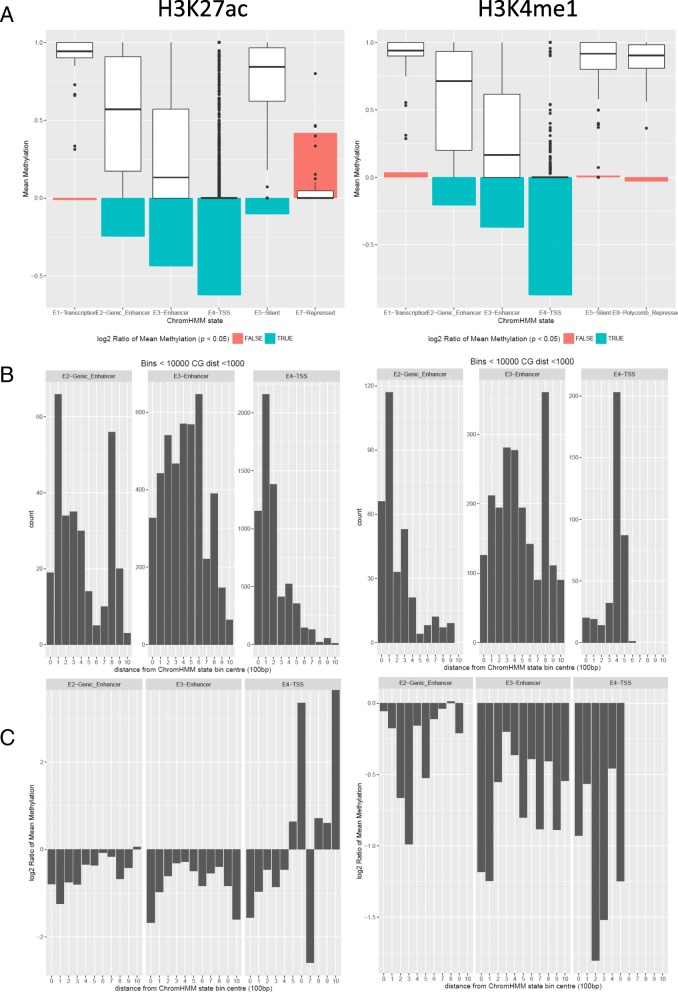
Results: We describe a large-scale coordinated study of allelic and non-allelic effects on DNA methylation, histone mark deposition, and gene expression, detecting the interrelations between epigenetic and functional features at unprecedented resolution. We use information from whole genome and targeted bisulfite sequencing from 910 samples to perform genotype-dependent analyses of allele-specific methylation (ASM) and non-allelic methylation (mQTL). In addition, we introduce a novel genotype-independent test to detect methylation imbalance between chromosomes. Of the ~2.2 million CpGs tested for ASM, mQTL, and genotype-independent effects, we identify ~32% as being genetically regulated (ASM or mQTL) and ~14% as being putatively epigenetically regulated. We also show that epigenetically driven effects are strongly enriched in repressed regions and near transcription start sites, whereas the genetically regulated CpGs are enriched in enhancers. Known imprinted regions are enriched among epigenetically regulated loci, but we also observe several novel genomic regions (e.g., HOX genes) as being epigenetically regulated. Finally, we use our ASM datasets for functional interpretation of disease-associated loci and show the advantage of utilizing naïve T cells for understanding autoimmune diseases.
Conclusions: Our rich catalogue of haploid methylomes across multiple tissues will allow validation of epigenome association studies and exploration of new biological models for allelic exclusion in the human genome.
Figures
References
-
- Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, Dilthey A, Ellis P, Langford C, Vannberg FO, Knight JC. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat Genet. 2012;44:502–10. doi: 10.1038/ng.2205. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
