

Functional Selectivity in Cytokine Signaling Revealed Through a Pathogenic EPO Mutation
- PMID: 28283061
- PMCID: PMC5376096
- DOI: 10.1016/j.cell.2017.02.026
Functional Selectivity in Cytokine Signaling Revealed Through a Pathogenic EPO Mutation
Abstract
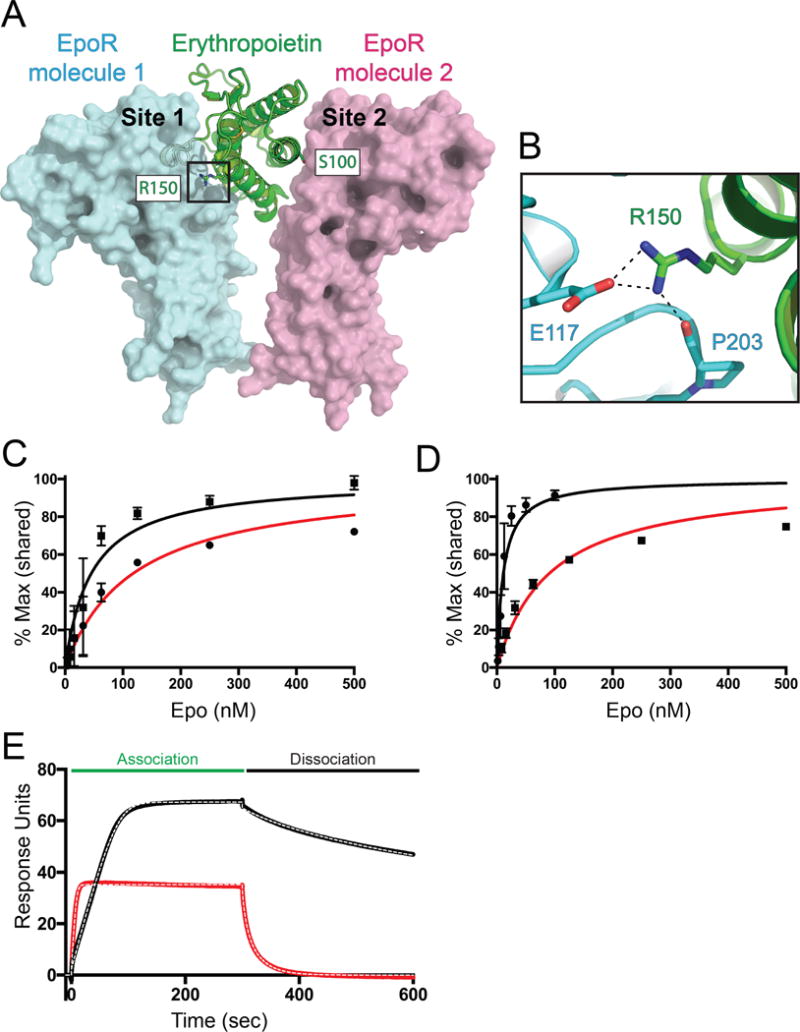
Cytokines are classically thought to stimulate downstream signaling pathways through monotonic activation of receptors. We describe a severe anemia resulting from a homozygous mutation (R150Q) in the cytokine erythropoietin (EPO). Surprisingly, the EPO R150Q mutant shows only a mild reduction in affinity for its receptor but has altered binding kinetics. The EPO mutant is less effective at stimulating erythroid cell proliferation and differentiation, even at maximally potent concentrations. While the EPO mutant can stimulate effectors such as STAT5 to a similar extent as the wild-type ligand, there is reduced JAK2-mediated phosphorylation of select downstream targets. This impairment in downstream signaling mechanistically arises from altered receptor dimerization dynamics due to extracellular binding changes. These results demonstrate how variation in a single cytokine can lead to biased downstream signaling and can thereby cause human disease. Moreover, we have defined a distinct treatable form of anemia through mutation identification and functional studies.
Keywords: JAK2; cytokine; erythropoiesis; erythropoietin; functional selectivity; hematopoiesis; receptor; signaling.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures






References
-
- Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, Wu J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70:562–570. - PubMed
-
- Chen Y, Wen R, Yang S, Schuman J, Zhang EE, Yi T, Feng GS, Wang D. Identification of Shp-2 as a Stat5A phosphatase. J Biol Chem. 2003;278:16520–16527. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
