

Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML
- PMID: 28283480
- PMCID: PMC5766841
- DOI: 10.1182/blood-2016-10-741207
Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML
Abstract
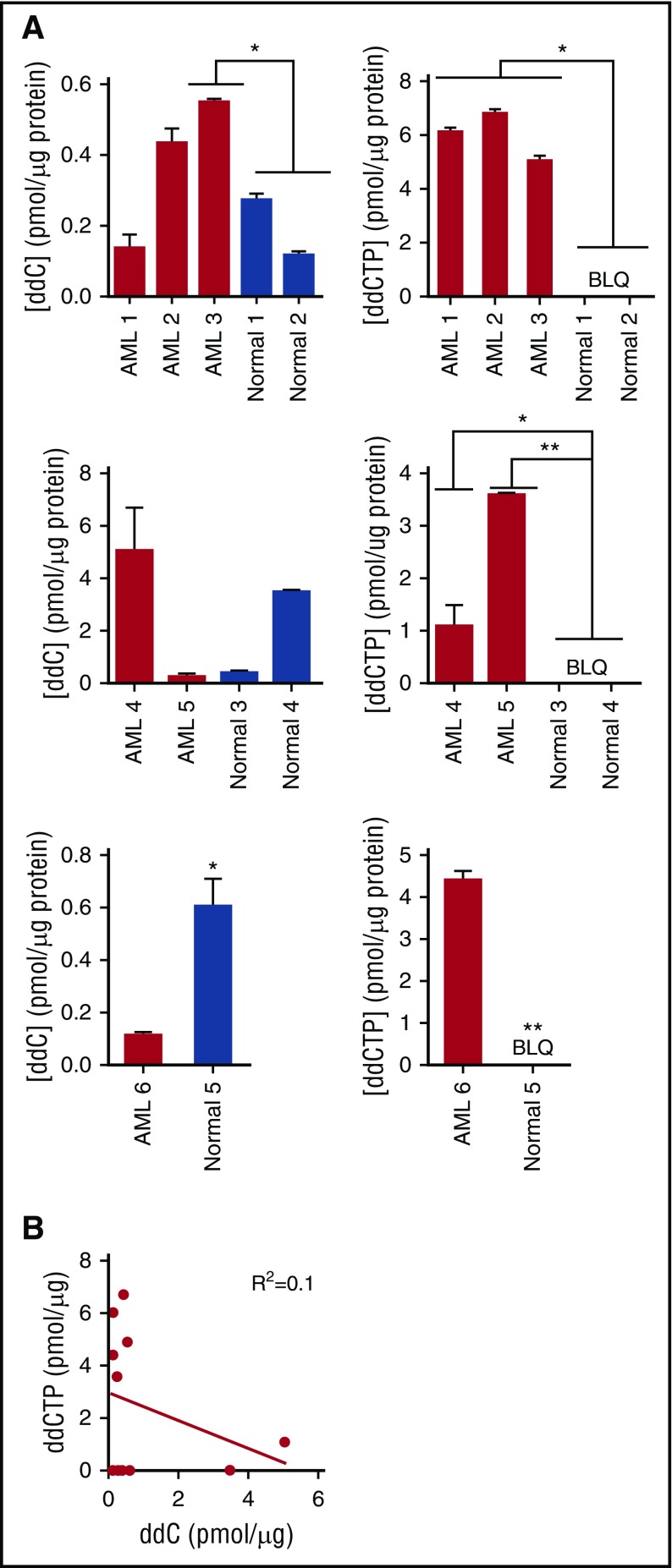
Mitochondrial DNA (mtDNA) biosynthesis requires replication factors and adequate nucleotide pools from the mitochondria and cytoplasm. We performed gene expression profiling analysis of 542 human acute myeloid leukemia (AML) samples and identified 55% with upregulated mtDNA biosynthesis pathway expression compared with normal hematopoietic cells. Genes that support mitochondrial nucleotide pools, including mitochondrial nucleotide transporters and a subset of cytoplasmic nucleoside kinases, were also increased in AML compared with normal hematopoietic samples. Knockdown of cytoplasmic nucleoside kinases reduced mtDNA levels in AML cells, demonstrating their contribution in maintaining mtDNA. To assess cytoplasmic nucleoside kinase pathway activity, we used a nucleoside analog 2'3'-dideoxycytidine (ddC), which is phosphorylated to the activated antimetabolite, 2'3'-dideoxycytidine triphosphate by cytoplasmic nucleoside kinases. ddC is a selective inhibitor of the mitochondrial DNA polymerase γ. ddC was preferentially activated in AML cells compared with normal hematopoietic progenitor cells. ddC treatment inhibited mtDNA replication, oxidative phosphorylation, and induced cytotoxicity in a panel of AML cell lines. Furthermore, ddC preferentially inhibited mtDNA replication in a subset of primary human leukemia cells and selectively targeted leukemia cells while sparing normal progenitor cells. In animal models of human AML, treatment with ddC decreased mtDNA, electron transport chain proteins, and induced tumor regression without toxicity. ddC also targeted leukemic stem cells in secondary AML xenotransplantation assays. Thus, AML cells have increased cytidine nucleoside kinase activity that regulates mtDNA biogenesis and can be leveraged to selectively target oxidative phosphorylation in AML.
© 2017 by The American Society of Hematology.
Figures






Comment in
-
A mitochondrial drug to treat AML.Blood. 2017 May 11;129(19):2597-2599. doi: 10.1182/blood-2017-03-773200. Blood. 2017. PMID: 28495924 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
