

Hypoxia-Mediated Mechanisms Associated with Antiangiogenic Treatment Resistance in Glioblastomas
- PMID: 28284719
- PMCID: PMC5417003
- DOI: 10.1016/j.ajpath.2017.01.010
Hypoxia-Mediated Mechanisms Associated with Antiangiogenic Treatment Resistance in Glioblastomas
Abstract
Glioblastomas (GBMs) are malignant tumors characterized by their vascularity and invasive capabilities. Antiangiogenic therapy (AAT) is a treatment option that targets GBM-associated vasculature to mitigate the growth of GBMs. However, AAT demonstrates transient effects because many patients eventually develop resistance to this treatment. Several recent studies attempt to explain the molecular and biochemical basis of resistance to AAT in GBM patients. Experimental investigations suggest that the induction of extensive intratumoral hypoxia plays a key role in GBM escape from AAT. In this review, we examine AAT resistance in GBMs, with an emphasis on six potential hypoxia-mediated mechanisms: enhanced invasion and migration, including increased expression of matrix metalloproteinases and activation of the c-MET tyrosine kinase pathway; shifts in cellular metabolism, including up-regulation of hypoxia inducible factor-1α's downstream processes and the Warburg effect; induction of autophagy; augmentation of GBM stem cell self-renewal; possible implications of GBM-endothelial cell transdifferentiation; and vasoformative responses, including vasculogenesis, alternative angiogenic pathways, and vascular mimicry. Juxtaposing recent studies on well-established resistance pathways with that of emerging mechanisms highlights the overall complexity of GBM treatment resistance while also providing direction for further investigation.
Copyright © 2017 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
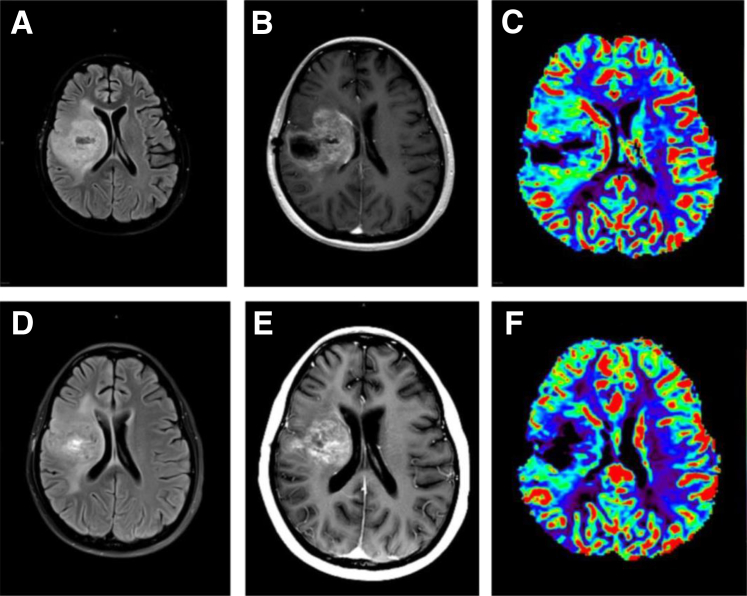
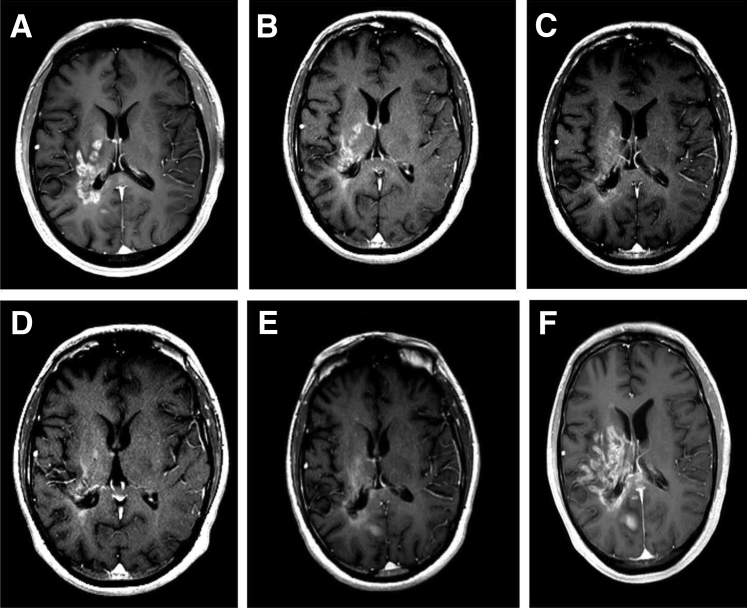
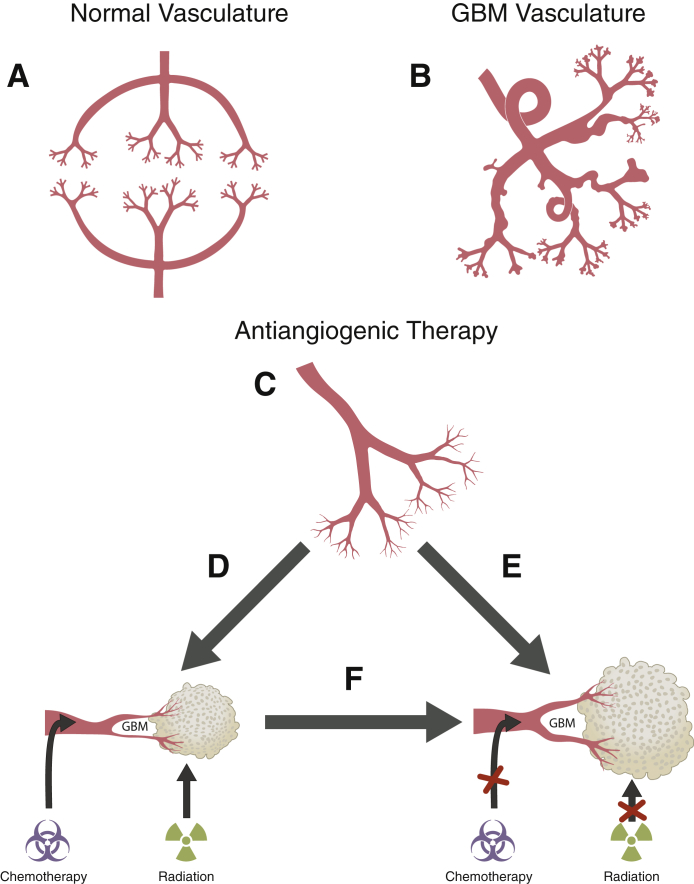
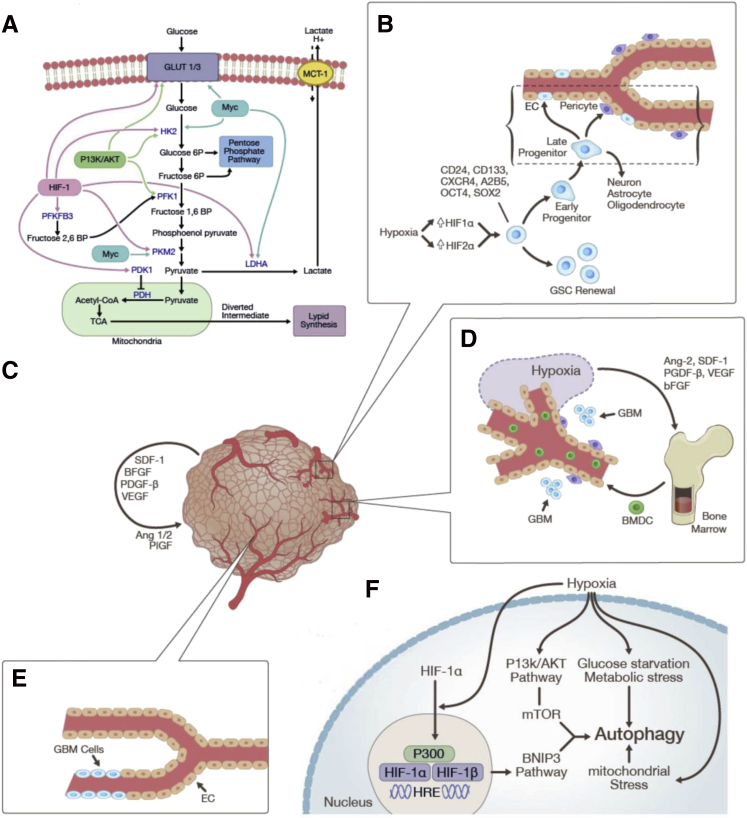
References
-
- Woehrer A., Bauchet L., Barnholtz-Sloan J.S. Glioblastoma survival: has it improved? Evidence from population-based studies. Curr Opin Neurol. 2014;27:666–674. - PubMed
-
- Wen P.Y., Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. - PubMed
-
- Gilbert M.R., Dignam J.J., Armstrong T.S., Wefel J.S., Blumenthal D.T., Vogelbaum M.A., Colman H., Chakravarti A., Pugh S., Won M., Jeraj R., Brown P.D., Jaeckle K.A., Schiff D., Stieber V.W., Brachman D.G., Werner-Wasik M., Tremont-Lukats I.W., Sulman E.P., Aldape K.D., Curran W.J., Jr., Mehta M.P. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. - PMC - PubMed
-
- Jain R.K., di Tomaso E., Duda D.G., Loeffler J.S., Sorensen A.G., Batchelor T.T. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
